
Analytical Instrument Design Qualification
Design Qualification establishes documented evidence that the selected analytical instrument and its supporting systems are suitable for the intended use defined in the User Requirements Specification. It is a pre-installation activity focused on confirming that the proposed design, configuration, and vendor-supplied solution will meet operational, performance, and regulatory expectations in a controlled laboratory environment.
Design Qualification bridges user requirements and actual system implementation. It ensures that procurement decisions are technically justified, risks are identified early, and that the instrument can be integrated into a compliant laboratory infrastructure without rework.
1. Objective of Design Qualification
The objective is to verify, through documented assessment, that:
- the instrument design aligns with defined analytical applications
- system capabilities meet critical performance requirements
- software and data handling functions support data integrity expectations
- supporting utilities and environmental conditions are compatible
- vendor design and documentation meet regulatory and quality standards
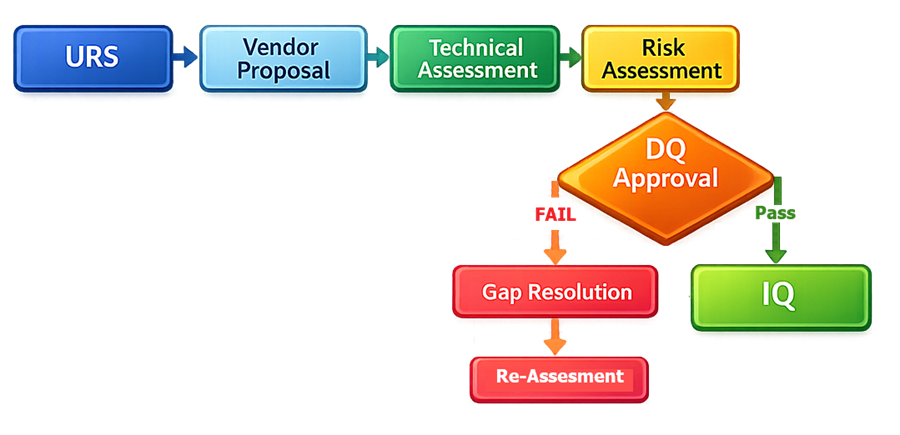
Design Qualification is not a test execution phase. It is a structured technical review supported by documented evidence. The diagram below illustrates the structured workflow used during Design Qualification. It shows how user requirements are systematically evaluated against vendor design, including technical assessment and risk evaluation, before formal approval to proceed to Installation Qualification. The workflow emphasizes the decision point where design suitability is confirmed or gaps are identified and resolved.
2. Inputs to Design Qualification
Design Qualification is driven by defined inputs. These must be complete and approved prior to execution.
Primary inputs include:
- approved User Requirements Specification
- vendor quotations and technical proposals
- instrument datasheets and specifications
- software functional descriptions
- factory acceptance test documentation if available
- applicable regulatory requirements such as 21 CFR Part 11
- internal standards for calibration, maintenance, and data integrity
Incomplete or vague inputs result in weak qualification and downstream deviations.
3. Design Review and Technical Assessment
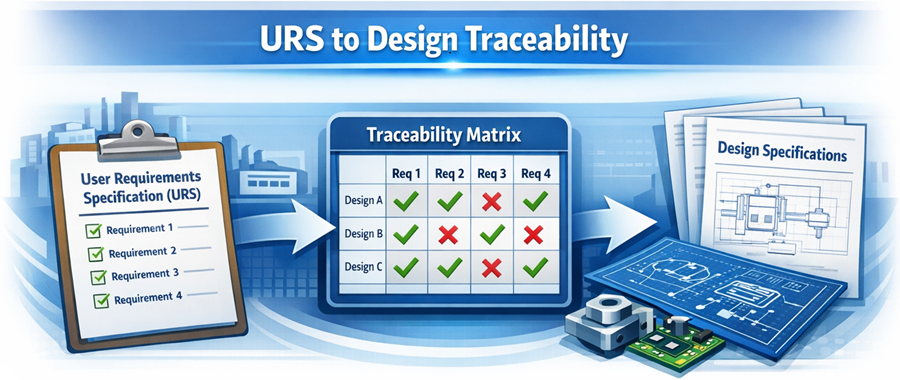
The core of Design Qualification is a structured technical review comparing user requirements against vendor design. The diagram below demonstrates the concept of requirement traceability during Design Qualification. Each user requirement is linked to a corresponding design feature or specification, ensuring that all intended functions are supported by the selected instrument configuration. Missing or unverified links represent gaps that must be addressed before qualification can proceed.
Key assessment areas include:
3.1 Instrument Hardware
- measurement range, sensitivity, and accuracy
- detector type and compatibility with intended methods
- sample introduction systems and throughput capability
- materials of construction and chemical compatibility
3.2 System Configuration
- module integration such as pumps, detectors, autosamplers
- scalability and upgrade capability
- compatibility with existing laboratory systems
3.3 Utilities and Infrastructure
- electrical requirements and power stability
- gas supply requirements where applicable
- HVAC and environmental conditions
- bench space and installation constraints
Each requirement in the URS must be traceable to a design feature or specification.
4. Software and Data Integrity Evaluation
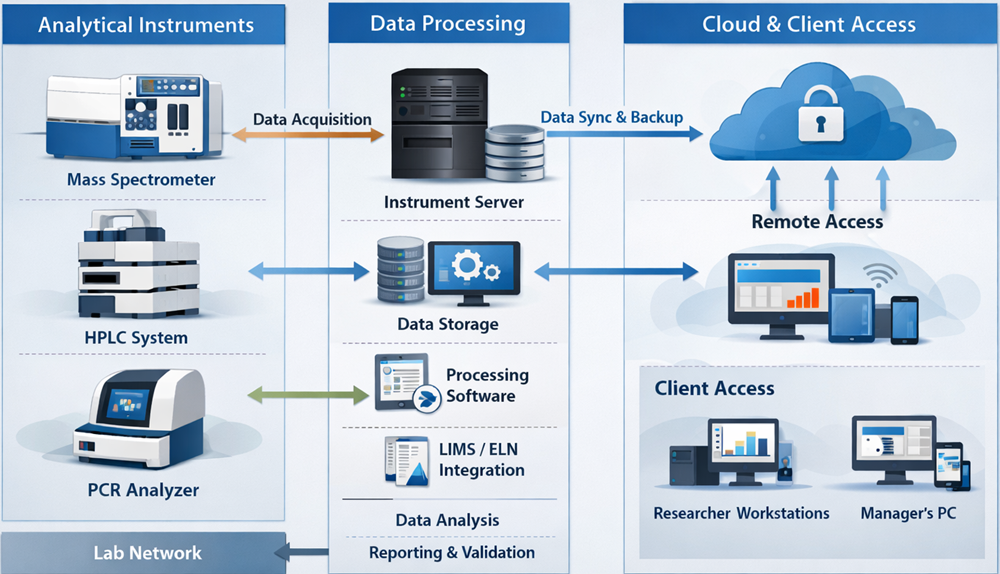
Analytical instruments frequently rely on computerized systems. Design Qualification must evaluate software capabilities in relation to regulatory expectations. The diagram below illustrates the typical architecture of an analytical instrument system, including hardware components, control software, data storage, and supporting IT infrastructure. It highlights how analytical data flows through the system from acquisition to storage and backup, providing context for evaluation of data integrity and system controls during Design Qualification.
Assessment includes:
- user access control and role-based permissions
- audit trail generation and review capability
- electronic data storage and protection
- data backup and recovery mechanisms
- compliance with 21 CFR Part 11 where applicable
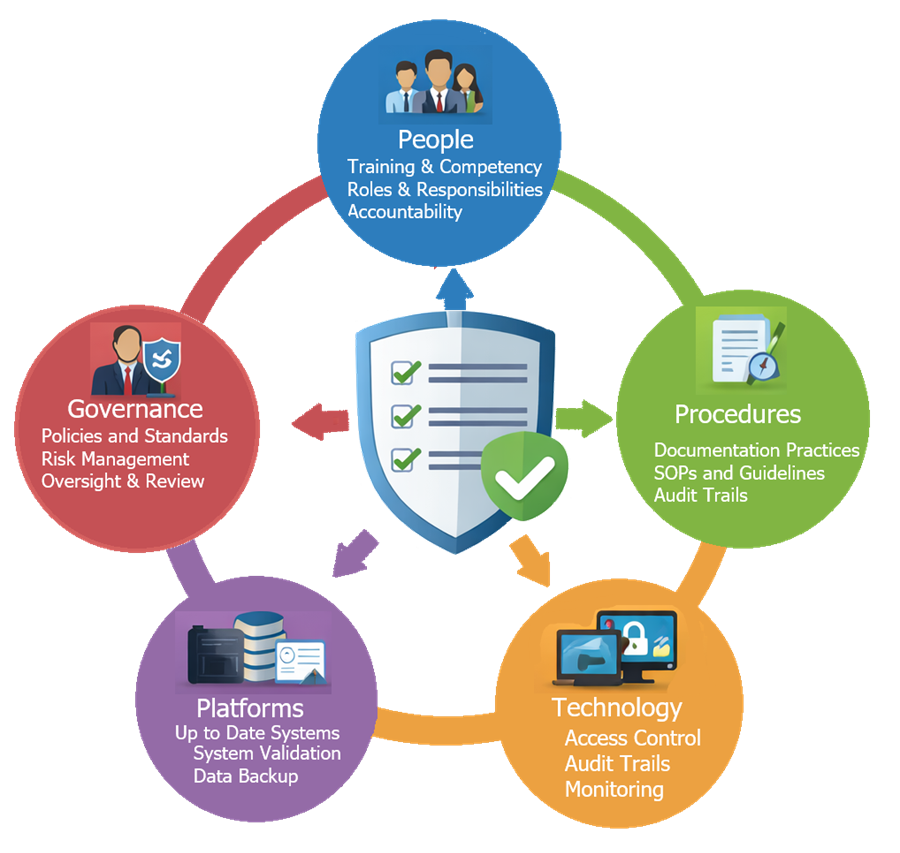
The diagram below presents the layered data integrity control framework evaluated during Design Qualification. It shows how user access, system controls, audit trails, and data storage mechanisms work together to protect electronic records and ensure compliance with regulatory expectations such as 21 CFR Part 11.
Gaps identified at this stage must be resolved before procurement or formally justified.
5. Vendor Assessment and Documentation Review
Vendor capability directly impacts validation success and must be evaluated as part of Design Qualification using a structured, evidence-based approach. The assessment should not be limited to document availability but must confirm that vendor deliverables are technically adequate, complete, and suitable for GMP use.
Vendor evaluation typically includes both organizational capability and documentation quality. From a capability standpoint, the vendor must demonstrate:
- established experience with regulated pharmaceutical or biotechnology environments
- ability to provide ongoing technical support, service, and spare parts
- defined processes for calibration, maintenance, and software lifecycle management
- responsiveness to deviations, issues, and change control requirements
From a documentation standpoint, Design Qualification must assess whether vendor-supplied materials can be leveraged within the validation program. This includes:
- Installation Qualification and Operational Qualification documentation, ensuring alignment with intended use and internal standards
- calibration certificates with traceability to recognized standards and defined uncertainty
- instrument specifications and functional descriptions that support requirement verification
- software validation documentation, including evidence of testing, version control, and change management
- maintenance procedures and recommended service intervals
Each document must be evaluated for:
- completeness relative to system configuration
- technical accuracy and internal consistency
- alignment with regulatory expectations such as 21 CFR Part 11 where applicable
- suitability for incorporation into GMP-controlled documentation
Where gaps are identified, they must be formally documented and addressed. Mitigation may include supplemental testing, development of internal documentation, or vendor clarification. Acceptance of vendor documentation without critical review introduces validation risk and is not acceptable within a compliant Design Qualification process.
6. Risk Assessment
A risk-based approach is applied to determine the level of qualification rigor required. The diagram below illustrates the risk-based approach used during Design Qualification. It shows how instrument complexity and impact on product quality are evaluated to determine the level of qualification rigor required. Higher risk systems require more extensive design review, testing, and documentation
Risk factors include:
- impact of the instrument on product quality decisions
- complexity of the instrument and software
- degree of automation and data processing
- detectability of failures
Risk assessment outputs drive:
- depth of testing during OQ and PQ
- need for additional controls
- frequency of requalification
7. Design Qualification Deliverables
Design Qualification must generate controlled, reviewable documentation that demonstrates a complete and objective evaluation of the proposed system design against defined user requirements. The deliverables must provide clear evidence that the selected instrument, configuration, and supporting systems are suitable for intended GMP use.
Design Qualification deliverables are not a collection of documents. They form an integrated body of evidence that supports a single conclusion: the design is acceptable or requires remediation.
Typical deliverables include:
- Design Qualification Protocol
Defines scope, responsibilities, acceptance criteria, and the structured approach used to perform the design review. It establishes how requirements will be evaluated and how conclusions will be documented. - Requirement-to-Design Traceability Matrix
Provides direct linkage between each user requirement and corresponding design feature, specification, or control. This document demonstrates coverage and identifies gaps or unverified requirements. - Documented Technical Assessment
Captures evaluation of instrument hardware, configuration, utilities, and system integration. This includes verification that performance capabilities, materials, and configuration align with intended analytical applications. - Risk Assessment Report
Documents the risk-based evaluation of the instrument, including impact on product quality, system complexity, and data integrity considerations. The output defines the level of qualification rigor and any required controls. - Vendor Evaluation Summary
Summarizes assessment of vendor capability and documentation, including suitability of supplied IQ/OQ materials, calibration traceability, software documentation, and service support. - Deviation and Gap Assessment with Resolution
Identifies all gaps between user requirements and design or documentation. Each gap must include documented impact assessment, defined mitigation, and final resolution prior to approval.
All Design Qualification deliverables must be reviewed and approved under document control by qualified personnel. The approval confirms that:
- all user requirements have been evaluated
- identified risks are understood and controlled
- gaps have been resolved or formally justified
- the system design is suitable to proceed to Installation Qualification
Design Qualification approval establishes the technical and compliance baseline for all subsequent qualification activities.
8. Common Deficiencies
Frequent failures in Design Qualification include:
- incomplete traceability between URS and design
- acceptance of vendor claims without verification
- insufficient evaluation of software and data integrity controls
- lack of defined intended use
- failure to assess integration with existing laboratory systems
These deficiencies result in rework during IQ and OQ and increase validation risk.
9. Relationship to Qualification Lifecycle
Design Qualification is positioned before Installation Qualification and defines the baseline for all subsequent testing.
- URS defines what is required
- DQ confirms the selected design can meet those requirements
- IQ verifies correct installation
- OQ verifies functional performance
- PQ confirms performance under routine conditions
Weak Design Qualification propagates defects through the entire lifecycle.
10. Documentation Expectations for Compliance
Design Qualification documentation must be:
- traceable to user requirements
- technically justified
- reviewed and approved by qualified personnel
- maintained under document control
The documentation must demonstrate that the instrument was selected based on objective, risk-based evaluation and is suitable for its intended GMP use.