
Container Closure Integrity Testing (CCIT)
1. Introduction
Container Closure Integrity Testing verifies that the sterile barrier created during aseptic processing remains intact throughout the product lifecycle. Sterility at time of fill does not ensure sterility at time of use. Integrity testing demonstrates that the container–closure system prevents microbial ingress, product leakage, and loss of barrier performance under storage and distribution conditions.
CCIT is therefore a sterility assurance control element integrated into validation lifecycle management.
2. Regulatory and Compendial Framework
CCIT expectations derive from cGMP requirements and pharmacopeial guidance.
U.S. Regulations
- 21 CFR 211.94 Drug Product Containers and Closures
- 21 CFR 211.113(b) Control of Microbiological Contamination
- 21 CFR 211.160(b) Laboratory Controls
Compendial and International Standards
- USP <1207> Package Integrity Evaluation – Sterile Products
- USP <1207.1> Deterministic Leak Test Technologies
- USP <1207.2> Microbial Ingress Test
- USP <1207.3> Package Seal Quality Test Technologies
- USP <382> Elastomeric Closure Functionality in Injectable Drug Packaging
- FDA Guidance for Industry – Sterile Drug Products Produced by Aseptic Processing
- EU GMP Annex 1 Manufacture of Sterile Medicinal Products
- ISO 11607 Packaging System Validation Principles
- PDA Technical Report No. 27 Pharmaceutical Package Integrity
Regulatory authorities increasingly expect scientifically justified deterministic methods when technically feasible.
3. Definition of Container Closure Integrity
Container Closure Integrity is the demonstrated ability of a container–closure system to maintain an effective sterile barrier throughout the product’s labeled shelf life. It extends beyond the condition of the seal at the time of filling and reflects sustained barrier performance under storage, transport, handling, and environmental stress conditions. An acceptable system must:
• Prevent microbial ingress
• Prevent product leakage or loss of internal pressure integrity
• Maintain seal performance over time
Sterility and integrity are distinct concepts. Sterility confirms absence of viable microorganisms at the time of testing. Integrity reflects the continued physical performance of the container–closure system to preserve that sterile state over time.
As illustrated in the Integrity Over Time diagram, barrier performance must be maintained from the point of sealing through storage, distribution, and the end of the labeled shelf life. Integrity therefore supports sustained sterility assurance, not merely initial release status.
CCIT therefore provides evidence that sterility assurance is maintained over time, not merely achieved at initial manufacture.
4. Sterile Barrier Systems and Potential Leak Paths
Common sterile barrier systems include:
• Vial–elastomeric stopper–aluminum crimp seal
• Pre-filled syringe–plunger stopper–tip cap or needle shield
• Blow-Fill-Seal containers
• Glass ampoules
• Cartridge systems
The cross-sectional diagram below illustrates typical sterile barrier interfaces and potential leak paths within a vial–stopper–crimp system.
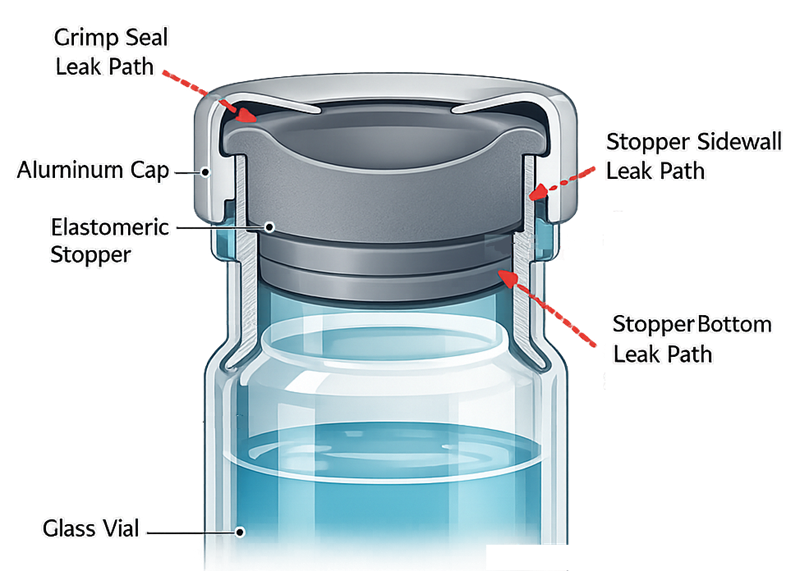
Typical leak mechanisms:
• Stopper–vial interface gaps
• Inadequate crimp compression
• Channel leaks
• Glass cracks or defects
• Elastomer imperfections
• Thermal expansion-induced seal disruption
Leak mechanism understanding must precede method selection.
5. CCIT Technologies
The diagram below compares deterministic and probabilistic integrity testing principles.
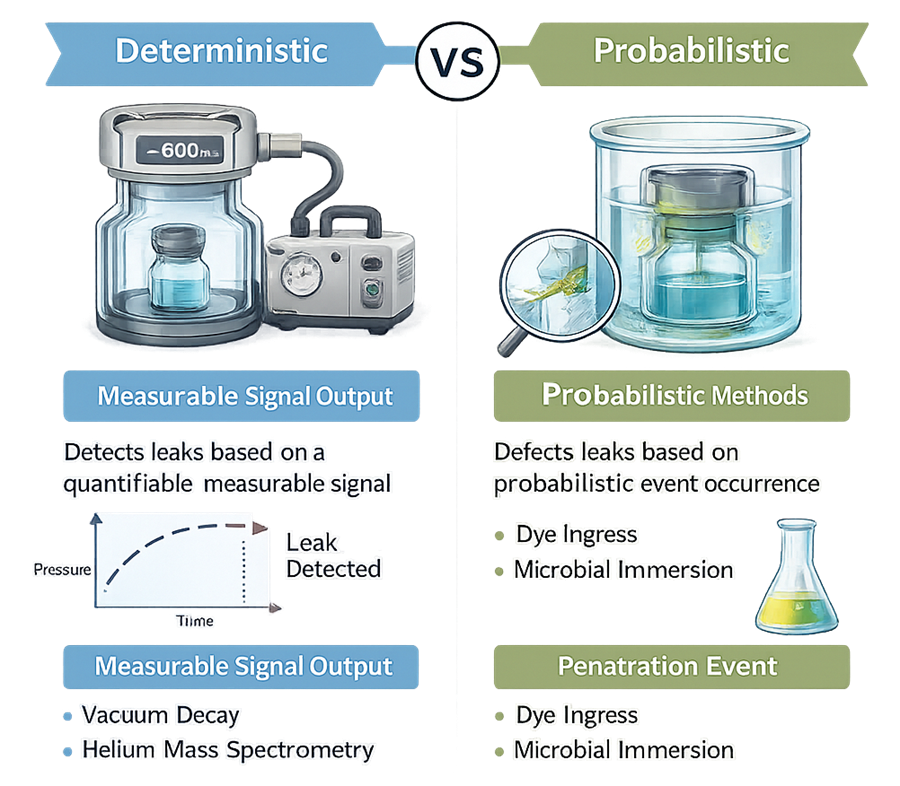
5.1 Deterministic Methods
Deterministic technologies detect leaks by measuring a physical parameter that changes in a predictable and quantifiable manner when a defect is present. The signal is based on fundamental principles such as pressure change, mass flow, electrical conductivity, or gas diffusion. The outcome does not rely on subjective interpretation or probabilistic penetration events.
A measurable, numerical response is generated and compared against a predefined acceptance threshold. This allows objective discrimination between intact and defective units. Common deterministic technologies include:
- Vacuum decay – Measures pressure rise in a test chamber evacuated around the package. A leak allows gas to escape from the container, producing a measurable pressure change.
- Pressure decay – Applies positive pressure and measures pressure loss over time due to leakage.
- Helium mass spectrometry – Detects helium escaping through micro-defects using highly sensitive mass detection. Suitable for very small leak sizes.
- High-voltage leak detection – Applies an electrical potential across a filled container. Current flow increases if a conductive path exists through a defect.
- Laser headspace analysis – Measures changes in headspace gas concentration or pressure using laser spectroscopy to detect leakage or seal compromise.
Vacuum decay testing detects leaks by measuring pressure rise within an evacuated chamber containing the test unit.
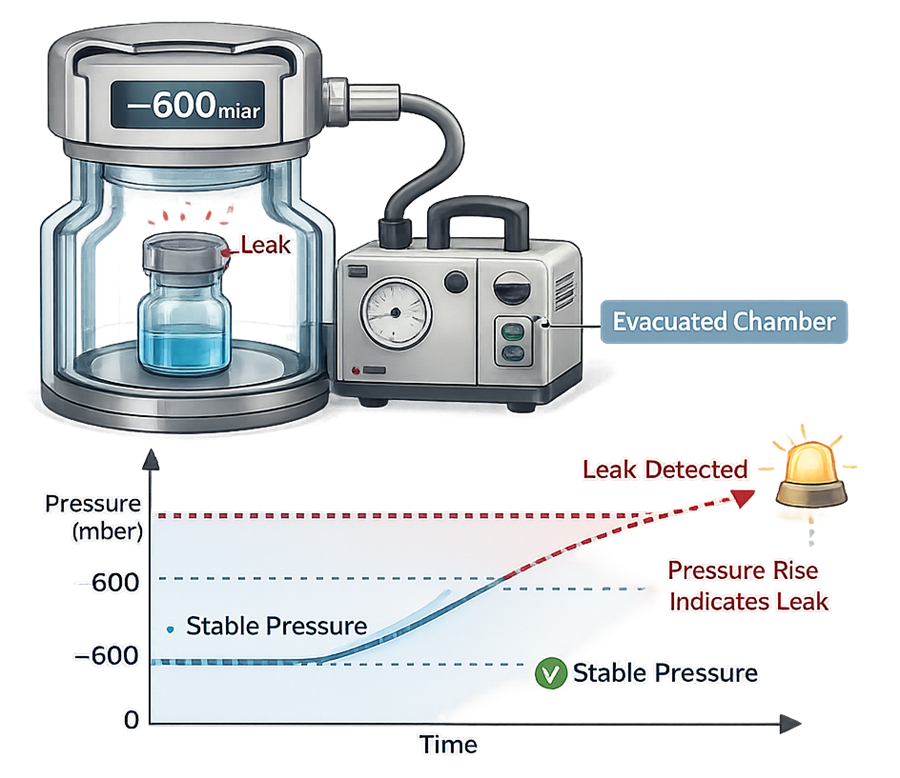
Key characteristics of deterministic methods:
- Quantitative output linked directly to defect presence
- High sensitivity capable of detecting sub-micron leak paths
- Predefined, objective pass/fail criteria
- Often non-destructive, allowing 100 percent inspection
- Strong statistical defensibility due to measurable response
Because these methods generate reproducible numerical data and can be validated against engineered defects, they provide a more robust and scientifically defensible assessment of container closure integrity compared to probabilistic techniques.
5.2 Probabilistic Methods
Probabilistic methods assess integrity by exposing the container–closure system to a challenge medium and observing whether penetration occurs. Detection depends on a stochastic event: the challenge agent must traverse the defect path under defined conditions. Absence of penetration does not prove absence of a defect; it only indicates that ingress did not occur under the specific test conditions.
Because the outcome depends on probability rather than direct physical measurement, these methods provide indirect evidence of integrity. Common probabilistic approaches include:
- Dye ingress – Units are submerged in a dye solution under vacuum or pressure conditions. After exposure, units are visually inspected for dye penetration through a defect path.
- Microbial ingress – Units are exposed to a microbial challenge suspension. Subsequent growth testing determines whether microorganisms penetrated the container–closure system.
Limitations of probabilistic methods:
- Lower sensitivity compared to deterministic technologies
- Detection dependent on test conditions such as pressure differential and exposure time
- Operator-dependent interpretation, particularly for visual dye detection
- Destructive testing, preventing 100 percent inspection
- Statistical uncertainty due to stochastic penetration events
Because these methods rely on probability rather than direct quantitative measurement, they offer reduced sensitivity and weaker statistical discrimination. For sterile injectable products, deterministic technologies are preferred when technically feasible and validated for the specific container–closure configuration.
6. Method Validation Strategy
Method validation must demonstrate that the selected CCIT technology can detect clinically relevant defects within the defined product–container configuration.
The validation logic below illustrates the structured sequence from engineered defect creation through routine lifecycle monitoring.
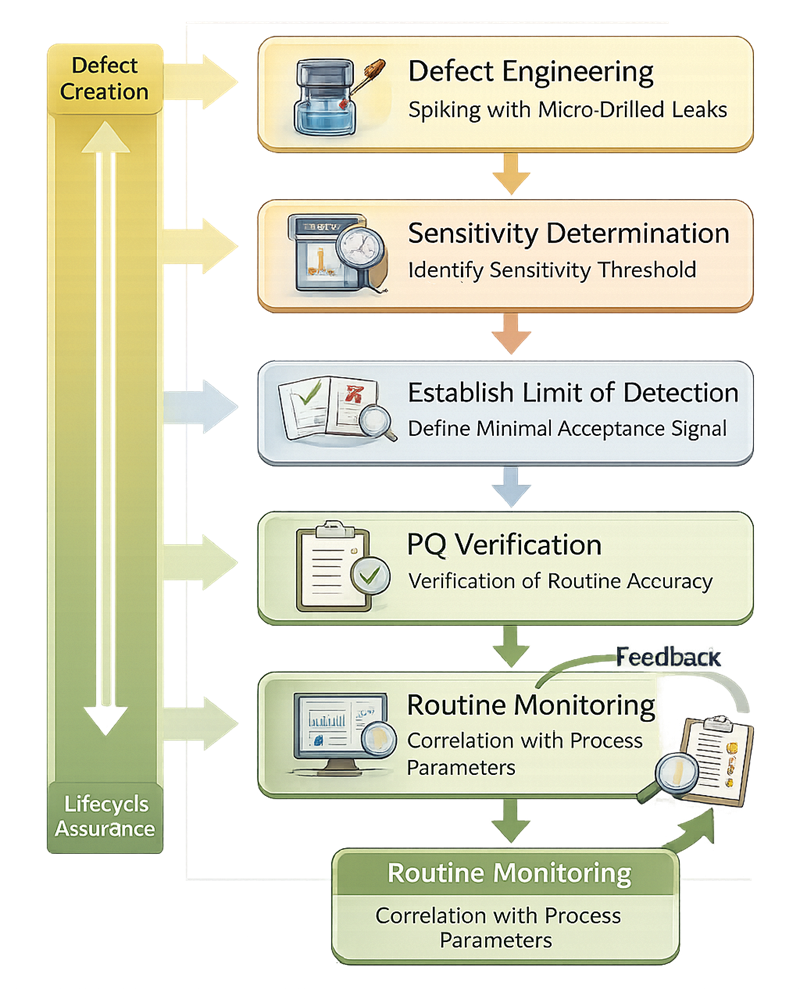
6.1 Sensitivity Determination
The smallest defect the method can detect must be demonstrated experimentally using engineered defects of known size. The selected defect size must be justified based on microbial ingress risk and product criticality. Sensitivity must be confirmed under worst-case conditions, including:
• Maximum fill volume
• Minimum headspace pressure differential
• Lowest acceptable stopper compression
• Temperature extremes
The method must reliably detect the defined defect at these operating limits to support a defensible detection claim.
6.2 Known Defect Creation and Characterization
Artificial defects must be intentionally created and dimensionally verified. Random damage is not acceptable. Acceptable approaches include:
• Laser-drilled microholes
• Precision capillary insertion
• Controlled stopper mis-seating
• Reduced crimp force studies
Each defect must be measured using calibrated metrology, documented, and uniquely traceable. The defect size and geometry must be known to support sensitivity validation.
6.3 Limit of Detection
The limit of detection must be established using statistically adequate sample sizes for each defined defect size. The probability of detection should be characterized relative to defect diameter.
The method must demonstrate clear discrimination between intact and defective units. Test results for acceptable units must not overlap with results from known defects at the defined detection limit.
6.4 False Positive and False Negative Assessment
Validation must quantify the frequency of incorrect results, including:
• Type I errors, where an intact unit is incorrectly identified as defective
• Type II errors, where a defective unit is incorrectly identified as intact
False negatives represent direct sterility risk and must be minimized and understood. Acceptance thresholds, retest rules, and disposition logic must be predefined in controlled procedures.
6.5 Repeatability and Reproducibility
System performance must be evaluated under normal operational variability, including:
• Multiple operators
• Multiple testing days
• Environmental variation within defined limits
• Multiple instruments where applicable
Statistical analysis must demonstrate consistent and reliable discrimination between intact and defective units across these conditions.
6.6 Worst-Case Product Configuration
If multiple container sizes, fill volumes, stopper types, or closure configurations exist, each configuration cannot be assumed equivalent. Either individual validation must be performed, or scientific grouping must be justified based on documented similarities such as geometry, headspace ratio, closure force, and material characteristics.
The worst-case configuration must be identified and documented. Worst-case typically reflects the condition most likely to reduce detection sensitivity or compromise seal integrity, such as lowest headspace, minimum stopper compression, or largest diameter interface.
Any change to container dimensions, elastomer formulation, crimp parameters, fill volume, or sealing equipment settings requires formal impact assessment. If the change alters the validated boundary conditions, partial or full revalidation may be required.
7. Sampling Strategy
Sampling must be risk-based and statistically justified.
7.1 Initial Validation Sampling
During Performance Qualification, sampling must be sufficient to demonstrate both the capability of the CCIT method and the consistency of the sealing process. The objective is not only to show that the test detects defects, but also that the manufacturing process produces an acceptably low defect rate under routine conditions.
Sampling expectations include:
• Elevated sample quantities – Sample size must be statistically justified and larger than routine monitoring levels. The intent is to generate meaningful confidence in process capability and detection performance during initial qualification.
• Beginning, middle, and end of lot representation – Samples must be taken throughout the filling run to capture potential variation due to equipment warm-up, mechanical drift, component feeding variation, or operator adjustment. Single-point sampling is not representative of batch performance.
• Multiple crimp heads and changeovers – If multiple sealing heads or stations are used, each must be represented in the validation dataset. Equipment changeovers, format adjustments, or maintenance events during the run must be included where applicable.
• Inclusion of seeded defect units – Known defective units must be intentionally introduced to confirm detection performance under actual production conditions. This verifies that the method maintains sensitivity when integrated into the validated process.
The overall objective of initial validation sampling is to demonstrate two elements simultaneously: The container closure process consistently produces units meeting integrity requirements across the full range of operational variability and the CCIT method can reliably detect predefined critical defects.
7.2 Stability Timepoint Testing
Integrity must be verified not only at release but throughout the product’s labeled shelf life. Testing at defined stability intervals confirms that the container–closure system maintains barrier performance over time under real storage and distribution conditions.
Stability evaluation should include:
- Initial timepoint – Establishes baseline integrity immediately after manufacture and provides reference data for comparison with later intervals.
- Intermediate storage – Confirms that seal performance remains stable during ongoing storage and detects early degradation trends.
- End-of-shelf-life – Demonstrates that the container–closure system maintains integrity through the full approved expiration period.
- Thermal cycling impact – Assesses the effect of temperature fluctuations that may occur during storage or transport, which can induce expansion, contraction, or seal relaxation.
- Transportation simulation effects – Evaluates mechanical stress such as vibration and shock that may affect closure integrity during distribution.
Maintenance of integrity over time is essential to sterility assurance. A system that is intact at release but degrades during storage does not meet regulatory expectations.
7.3 Ongoing Production Monitoring
Routine monitoring requirements must be defined in controlled SOPs and supported by documented justification. Sampling frequency and acceptance criteria should reflect actual process performance and product risk.
The monitoring strategy should be based on:
- Historical defect trends – Review of prior CCIT data to understand defect frequency, variability, and emerging patterns.
- Process capability – Demonstrated stability and statistical capability of the sealing process. Higher capability may justify optimized sampling; unstable processes require increased oversight.
- Product risk classification – Consideration of route of administration, patient population, and clinical impact of potential failure.
Trending is mandatory. Borderline or near-threshold results must be evaluated for drift or degradation patterns. CCIT data should also be correlated with critical equipment parameters such as crimp force, stopper compression, and maintenance history to detect early signals of process deterioration.
7.4 Risk-Based Lot Sampling
Sampling intensity must be risk-based and proportionate to the potential clinical impact of failure. The level of inspection should increase as product risk increases or as process uncertainty rises.
Sampling strategy should consider:
- Route of administration – Parenteral products, especially intrathecal or ophthalmic, require more conservative sampling due to direct systemic exposure.
- Patient risk profile – Products intended for immunocompromised or critical-care patients warrant tighter control.
- Container size and configuration – Larger interfaces or complex geometries may present higher leak risk.
- Historical performance – Stable processes with demonstrated low defect rates may justify optimized sampling; adverse trends require increased sampling.
- Maintenance events – Recent crimp head servicing or equipment adjustment may temporarily elevate risk.
- Change control activities – Component changes, supplier changes, or process parameter adjustments require reassessment of sampling adequacy.
For non-destructive deterministic systems capable of high-sensitivity inspection, traditional AQL-based attribute sampling is generally inappropriate because the technology can support enhanced or 100 percent inspection strategies.
If destructive probabilistic methods are used, statistically justified acceptance sampling plans must be defined, documented, and scientifically defended, including confidence level and acceptable defect rate assumptions.
8. Integration with Aseptic Filling and Sealing Validation
CCIT cannot function as an isolated laboratory test. It must be technically aligned with the mechanical and process parameters that create the sterile barrier. Integrity results are meaningful only when linked to validated sealing conditions.
CCIT must align with:
- Stopper compression qualification – Compression range must be established during equipment qualification to ensure adequate seal formation without over-compression. CCIT verifies that the validated compression window consistently produces acceptable integrity performance.
- Crimp force validation – Crimp diameter, roll depth, and skirt formation must be defined and controlled. CCIT data should correlate with validated crimp settings to confirm that mechanical closure parameters support barrier integrity.
- Line speed qualification – Maximum validated line speed must not compromise stopper seating or crimp consistency. CCIT during PQ should include worst-case speed conditions to confirm seal robustness under dynamic production stress.
- Component dimensional tolerance studies – Variability in vial neck finish, stopper dimensions, and aluminum seal geometry must be evaluated during qualification. CCIT performance must remain acceptable across defined component tolerance limits.
- Preventive maintenance programs – Crimp heads, chuck components, and tooling wear directly influence seal formation. Maintenance intervals must be justified using integrity performance data and trending.
CCIT data should be periodically reviewed alongside mechanical parameter trends. A shift in crimp diameter distribution or stopper compression force without a corresponding change in integrity performance may indicate early process drift.
Any change to sealing parameters, component suppliers, elastomer formulation, container geometry, line configuration, or tooling requires formal impact assessment. If the change alters validated boundary conditions or worst-case assumptions, partial or full revalidation of CCIT may be required.
CCIT is therefore an integrated control element within the aseptic filling validation framework, not a stand-alone laboratory exercise.
9. Lifecycle Control and Revalidation Triggers
CCIT is not limited to initial qualification. Integrity assurance must be maintained through structured lifecycle oversight consistent with contamination control strategy and process validation governance.
Ongoing lifecycle management must include:
- Trending of integrity data – Routine review of acceptance rates, borderline values, and detection signal distribution to identify drift or emerging patterns.
- Investigation of deviations – Formal root cause analysis of failures, repeated borderline results, or unexpected variability. Corrective and preventive actions must address both equipment and component factors.
- Review during Annual Product Quality Review – Periodic evaluation of integrity performance, defect rates, and correlation with sealing parameters as part of product quality trending.
- Assessment following component changes – Impact evaluation when vial, stopper, seal, or supplier changes occur. Scientific justification must confirm that validated boundary conditions remain applicable.
- Evaluation after equipment modification – Reassessment following tooling replacement, crimp head servicing, automation upgrades, or parameter adjustment.
Revalidation may be required when changes affect the validated sealing envelope or detection assumptions, including:
- Component formulation or dimensional change
- Crimp head replacement or mechanical modification
- Repeated borderline or unexplained failures
- Sustained process parameter drift outside established control ranges
Integrity performance must remain within validated limits over time. CCIT therefore functions as a continuous sterility assurance control embedded within the overall validation lifecycle and contamination control strategy.