
EtO Sterilization Validation
Ethylene oxide sterilization validation demonstrates that a defined sterilization process consistently achieves the required Sterility Assurance Level under worst-case, controlled conditions.
Because EtO sterilization is a multi-variable chemical process involving gas concentration, humidity, temperature, time, and load configuration, validation must address both microbiological lethality and chemical residual safety. The process cannot be qualified based on chamber parameters alone.
Validation must be science-based, risk-aligned, and lifecycle-controlled.
1. Regulatory and Standards Framework
EtO sterilization validation is primarily governed by:
- ISO 11135 — Sterilization of health care products — Ethylene oxide
- ISO 10993-7 — Ethylene oxide sterilization residuals
- FDA 21 CFR Part 820 for medical devices
- FDA 21 CFR Parts 210 and 211 for pharmaceuticals where applicable
Regulators expect:
- Documented development studies
- Defined worst-case conditions
- Demonstrated reproducibility
- Ongoing process monitoring
- Controlled change management
Validation is not a single protocol. It is a structured lifecycle.
2. Validation Strategy Overview
EtO sterilization validation is managed as a controlled lifecycle process integrating development, qualification, routine monitoring, and requalification.
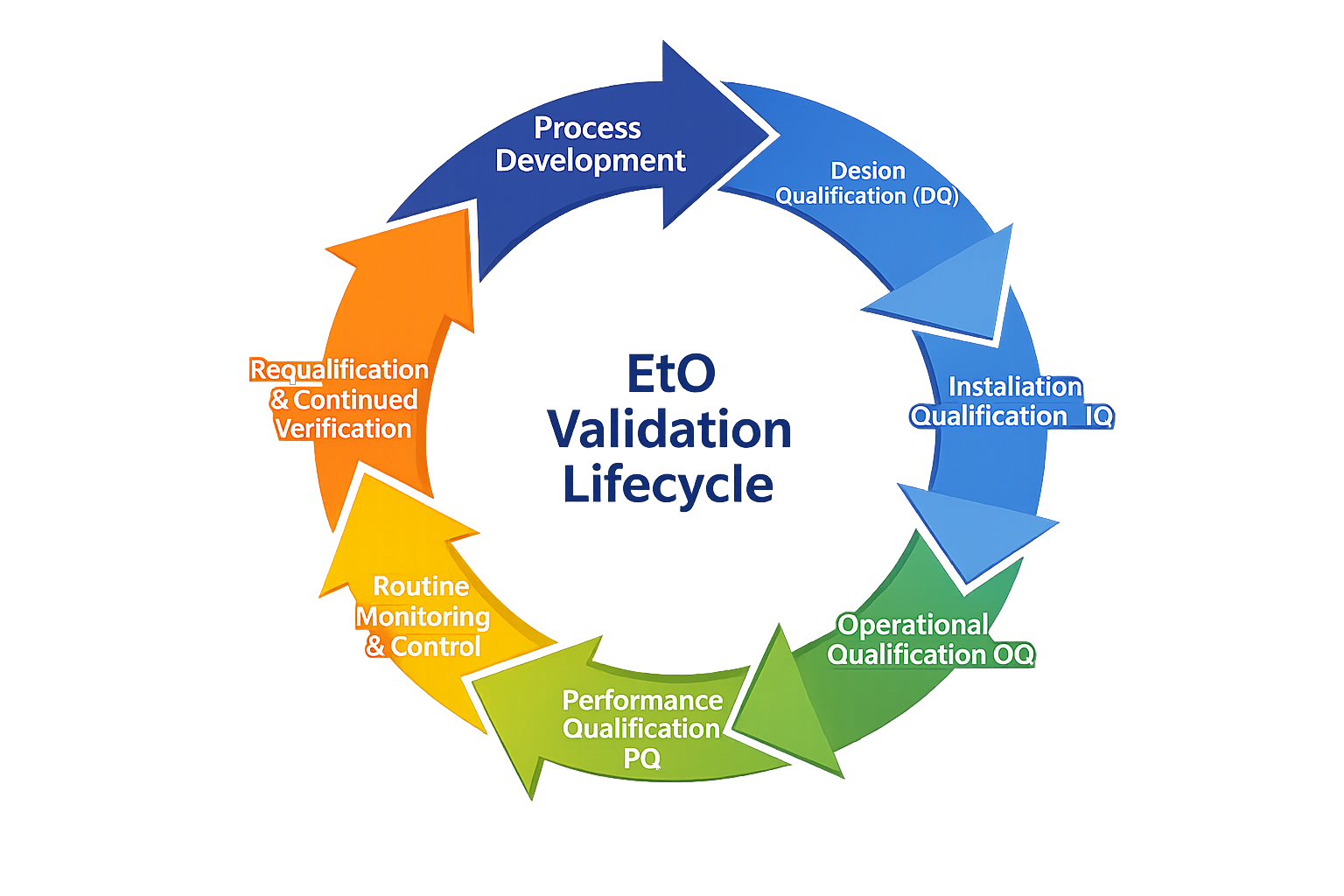
EtO validation typically follows a staged approach:
- Process Development
- Installation Qualification IQ
- Operational Qualification OQ
- Performance Qualification PQ
- Routine Monitoring and Control
- Requalification and Continued Verification
Each stage builds on defined risk assessment and scientific rationale.
3. Process Development
Process development establishes:
- Target temperature
- Relative humidity range
- Gas concentration
- Exposure time
- Load configuration
- Aeration parameters
Development activities may include:
- Half-cycle lethality studies
- Biological indicator placement mapping
- Worst-case lumen evaluation
- Residual aeration studies
- Parametric sensitivity assessment
The goal is to define a robust operating window before formal qualification begins. Poor development leads to unstable validation.
4. Installation Qualification IQ
IQ verifies that the sterilization system and supporting infrastructure are installed according to approved design specifications. Typical IQ elements include:
- Chamber construction verification
- Vacuum system verification
- Gas delivery system verification
- Humidity injection verification
- Control system configuration
- Safety interlocks
- Emission control systems
- Utility connections
IQ confirms the system is built correctly.
5. Operational Qualification OQ
OQ demonstrates that the sterilizer operates within defined parameter ranges under empty and controlled load conditions.
OQ typically evaluates:
- Temperature distribution
- Humidity control performance
- Gas injection accuracy
- Pressure and vacuum performance
- Alarm functionality
- Interlock performance
Biological indicators may be used during OQ to confirm lethality capability under defined conditions. Worst-case parameter combinations must be challenged.
6. Performance Qualification PQ
PQ demonstrates consistent sterilization performance under routine production load conditions.
PQ typically includes:
- Defined worst-case load configuration
- Biological indicator placement in most challenging locations
- Minimum of three consecutive successful cycles
- Half-cycle or overkill validation approach where applicable
- Residual testing to confirm compliance with ISO limits
PQ confirms that the validated cycle achieves the required Sterility Assurance Level in actual product configuration. This is the critical regulatory milestone.
7. Biological Indicator Strategy
EtO validation commonly uses spores of Bacillus atrophaeus due to their resistance characteristics. The half-cycle validation model is based on demonstrated logarithmic reduction of biological indicator organisms over exposure time, establishing a defined lethality margin.
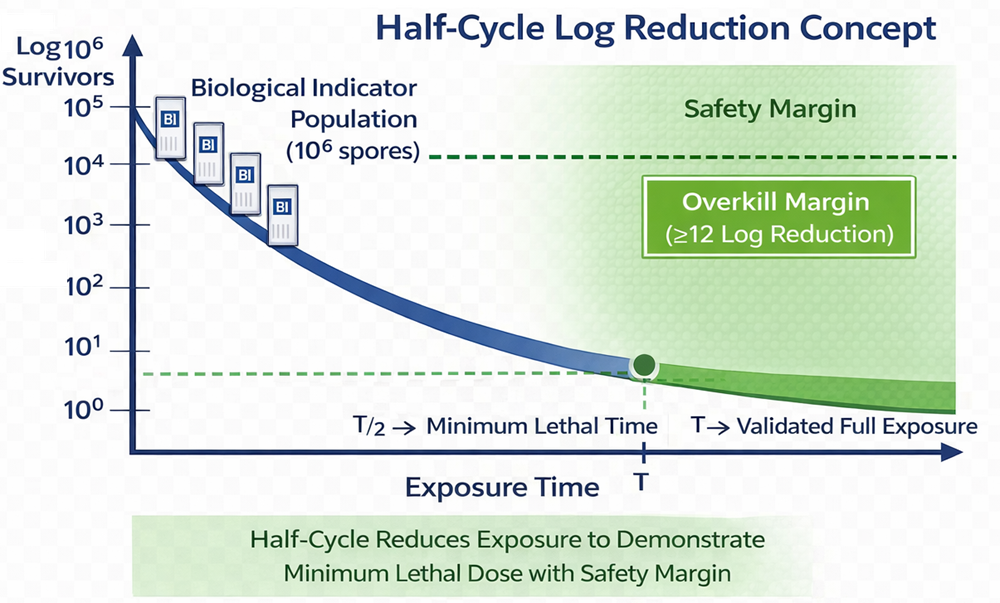
Key considerations:
- BI population and resistance verification
- Placement in worst-case locations
- Lumen simulation where applicable
- Half-cycle demonstration
- Fractional cycle confirmation
BI use must align with selected validation method.
Biological indicators must be positioned in the most difficult-to-sterilize locations within the defined worst-case load configuration, as illustrated below.
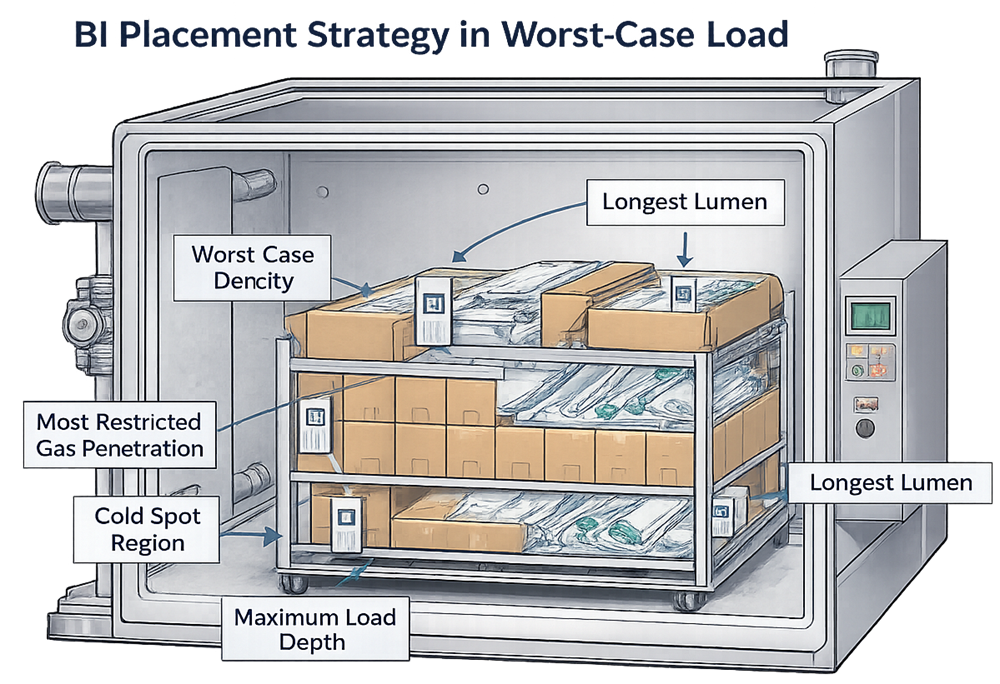
8. Residual Evaluation and Aeration Validation
Validation must demonstrate that residual EtO and byproducts remain within acceptable limits. This includes:
- Aeration time justification
- Temperature-controlled aeration studies
- Residual extraction and analytical testing
- Toxicological risk assessment
Sterility without residual compliance is not acceptable.
9. Routine Monitoring and Control
After PQ, the process enters controlled production status. Routine monitoring typically includes:
- Recorded cycle parameters
- Gas injection quantity verification
- Temperature and humidity trending
- BI periodic confirmation
- Residual verification as required
- Defined release criteria
Data integrity and traceability are critical.
10. Requalification and Continued Verification
Requalification may be triggered by:
- Equipment modification
- Gas supply changes
- Software updates
- Load configuration changes
- Process drift
- Extended downtime
The scope of requalification must reflect risk and historical performance. Document review alone is acceptable only when risk is demonstrably low. Requalification decisions must be based on structured impact assessment and defined risk evaluation logic, as illustrated below.
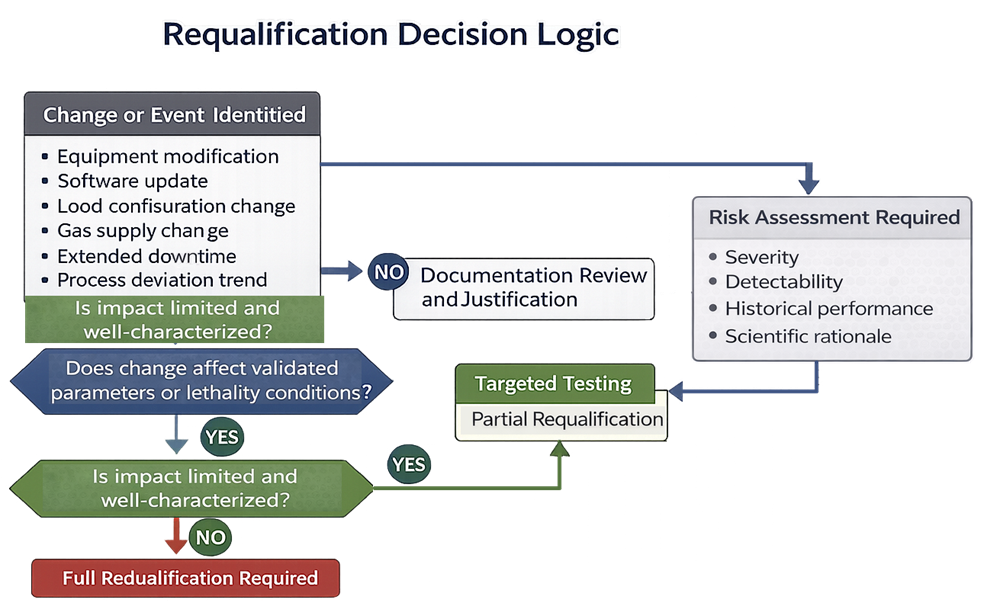
Validation Lifecycle Integration
EtO sterilization validation is a structured, risk-based lifecycle process integrating microbiology, chemical safety, engineering controls, and regulatory compliance. It requires disciplined development, rigorous qualification, and ongoing monitoring to maintain a validated state. Failure in any of these areas commonly results in regulatory observations.