
Media Fill and Aseptic Process Simulation
Media fill, or aseptic process simulation, is the microbiological verification of an aseptic manufacturing process. It demonstrates that the integrated filling line, operating environment, barrier system, and personnel practices collectively maintain sterility under defined worst-case conditions.
Unlike mechanical qualification, which verifies equipment function, media fill challenges the entire aseptic process using sterile growth medium in place of product. Its purpose is to confirm that no microbial contamination is introduced during routine or simulated non-routine operations.
The diagram below presents the structured sequence of a media fill as a controlled, closed-loop verification process. It illustrates progression from media preparation through aseptic simulation, incubation, inspection, and final contamination decision logic. This model emphasizes that media fill is a defined validation exercise with predefined decision criteria, not merely a broth-filling activity.
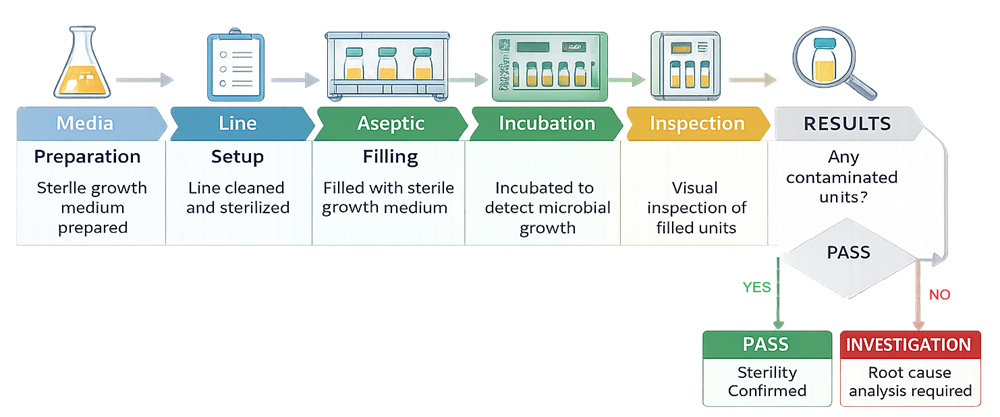
1. Objective and Regulatory Context
The objective of media fill is to provide documented evidence that the aseptic process can consistently produce sterile units when operated within validated limits. Media fill is required for:
• Initial qualification of a new aseptic filling line
• Introduction of new container formats
• Significant equipment modification
• Major facility changes
• Requalification following high-risk deviations
It is not a substitute for equipment qualification. It confirms microbiological performance after the system has been mechanically and operationally qualified.
2. Scope of Simulation
Media fill must simulate:
• Actual production setup
• Maximum planned interventions
• Routine and non-routine operator actions
• Longest anticipated batch duration
• Maximum line speed where applicable
• Representative container sizes
The simulation must reflect worst-case conditions rather than ideal production scenarios. For lines connected to lyophilizers, the simulation must include transfer and partial stoppering steps.
3. Selection of Growth Medium
Tryptic Soy Broth is commonly used due to its ability to support growth of a wide range of microorganisms. The selected medium must:
• Be sterile prior to use
• Support growth promotion testing
• Be compatible with container and closure system
• Be visually suitable for turbidity detection
Growth promotion testing must confirm that the medium supports microbial growth under incubation conditions.
4. Batch Size and Duration
Batch size must be statistically representative and aligned with regulatory expectations. The number of units filled should reflect routine commercial scale where possible. The duration must represent:
• Longest planned exposure time
• Shift changes
• Maximum intervention frequency
Extended runs challenge fatigue, intervention frequency, and environmental stability.
5. Interventions and Operator Simulation
Interventions represent the highest contamination risk during aseptic processing. Media fill must therefore deliberately challenge the process by incorporating predefined routine and non-routine interventions under realistic operating conditions.
The Intervention Simulation Matrix below defines the required structure for intervention inclusion. It categorizes intervention types, assigns relative contamination risk, and specifies inclusion requirements for both initial qualification and periodic requalification. It also defines the expected simulation approach for each category.
Routine interventions such as component replenishment, glove manipulation, or standard line clearance must be represented in every media fill because they occur in normal production. Medium- and high-risk interventions must be executed under defined worst-case conditions, including maximum speed, extended exposure duration, or post-adjustment restart where applicable.
Non-routine interventions, including mechanical adjustments or barrier access events, must be included during initial qualification and reintroduced during requalification based on documented risk assessment and historical performance data.
All interventions must be predefined in the approved protocol. Frequency, timing, and execution method must be documented prior to execution. Selective omission or reduction of high-risk interventions to improve outcome probability is not acceptable.
Operator technique and aseptic discipline are evaluated indirectly through contamination outcome and environmental monitoring results associated with the intervention categories defined in the matrix below.
| Intervention Type | Example Activity | Risk Level | Required During Initial Media Fill | Required During Periodic Requalification | Simulation Approach |
|---|---|---|---|---|---|
| Routine – Minor | Glove adjustment within ISO 5 | Low | Yes | Yes | Performed at defined frequency |
| Routine – Component Replenishment | Stopper or cap replenishment | Medium | Yes | Yes | Performed at worst-case exposure duration |
| Routine – Line Clearance | Removal of minor jam | Medium | Yes | Yes | Simulated under operating speed |
| Non-Routine – Mechanical Adjustment | Needle height adjustment | High | Yes | Risk-based | Simulated under controlled but realistic conditions |
| Non-Routine – Sensor Intervention | Clearing misread container | Medium to High | Yes | Risk-based | Performed during active filling |
| Non-Routine – Extended Door Opening | Barrier access event | High | Yes | Risk-based | Simulated with defined exposure time |
| Startup / Shutdown Transition | Restart after pause | Medium | Yes | Yes | Included in long-duration runs |
| Shift Change Activity | Operator handover | Medium | Yes | Yes | Simulated with full gown transition |
Decision Principles
- Low-risk routine interventions must always be represented because they occur in every batch.
- Medium-risk interventions must be included at defined frequency and under operational speed.
- High-risk or non-routine interventions must be included in initial qualification and reintroduced during periodic requalification based on risk assessment and historical performance.
This matrix ensures that media fill remains a realistic contamination challenge rather than a minimized demonstration.
6. Environmental Monitoring Integration
Environmental monitoring must be conducted during media fill under dynamic conditions. This includes:
• Viable air sampling
• Surface sampling
• Glove fingertip monitoring
• Non-viable particle monitoring
Data from environmental monitoring supports investigation if contaminated units are detected.
7. Incubation and Visual Inspection
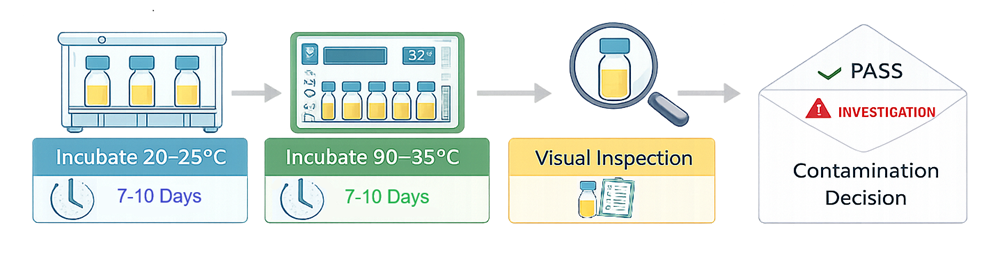
Following completion of the aseptic simulation, filled units are subjected to controlled incubation to promote detection of microbial growth. Incubation is a defined and validated step of the media fill process and must be performed under controlled and documented conditions.
Incubation Strategy
Incubation is typically performed using a dual-temperature sequence to promote recovery of a broad spectrum of microorganisms. A common approach includes:
• Initial incubation at 20–25°C for 7–10 days
• Subsequent incubation at 30–35°C for 7–10 days
The sequence may be reversed depending on established site procedure, but total incubation duration generally ranges from 14 to 20 days. The defined temperatures and duration must be specified in the approved protocol prior to execution. Incubation conditions must ensure:
• Uniform temperature distribution within the incubator
• Controlled and monitored temperature setpoints
• Documented temperature recording
• Defined loading configuration to avoid airflow obstruction
Overloading or improper arrangement of units may compromise temperature uniformity and detection sensitivity.
Incubators State
Incubators used for media fill evaluation must be in a qualified and controlled state. This includes:
• Installation and operational qualification
• Temperature mapping to demonstrate uniformity
• Calibrated temperature sensors
• Alarm verification
• Preventive maintenance under change control
Use of unqualified or out-of-calibration incubators invalidates media fill results. Incubation is a critical verification step and must meet the same lifecycle control standards as production equipment.
Visual Inspection
Following incubation, each unit must be individually inspected for evidence of microbial growth. Inspection is typically performed against a suitable light source with contrasting background to enhance detection of turbidity or particulate growth. Visual inspection must assess:
• Turbidity of the growth medium
• Presence of visible colonies
• Sediment formation
• Surface pellicle development
• Gas production where applicable
Inspection conditions must be controlled and consistent. Personnel performing inspection must be trained and qualified. Inspection criteria must be predefined and documented in the protocol. Any suspect unit must be segregated and evaluated according to investigation procedures. Confirmatory testing may be required to differentiate true microbial growth from cosmetic artifacts such as air bubbles or particulate contamination.
Incubation and inspection collectively represent the microbiological detection phase of media fill. These steps convert the aseptic simulation from mechanical demonstration into microbiological evidence of sterility assurance.
8. Acceptance Criteria and Evaluation
Acceptance criteria for media fill must be predefined in the approved protocol prior to execution. Criteria must be aligned with applicable regulatory guidance and reflect batch size, production scale, and risk profile of the aseptic process.
Acceptance criteria are not limited to the presence or absence of contaminated units. They must define quantitative thresholds, decision pathways, and mandatory investigation triggers.
The table below provides a structured acceptance framework.
| Parameter | Defined Requirement | Regulatory Consideration | Action if Not Met |
|---|---|---|---|
| Total Units Filled | Predefined batch size representative of routine production | Must reflect worst-case duration and intervention frequency | Media fill invalid if insufficient units |
| Number of Contaminated Units | Zero growth expected for typical batch sizes | Zero tolerance commonly expected for ≤ 5,000 units | Immediate investigation required |
| 1 Contaminated Unit (Small Batch) | Not acceptable | Considered process failure in most cases | Full investigation and repeat media fill |
| 1 Contaminated Unit (Large Batch) | Evaluate per regulatory threshold | Statistical guidance may apply for very large batches | Investigation; risk-based decision on repeat |
| ≥ 2 Contaminated Units | Automatic failure | Indicates loss of sterility control | Process considered failed; requalification required |
| Growth Promotion Test | Must demonstrate medium supports microbial growth | Required to validate detection capability | Media fill invalid if GPT fails |
| Environmental Monitoring During Run | Within predefined alert/action limits | Supports investigation context | Exceedance requires evaluation |
| Intervention Execution | All predefined interventions completed | Must represent worst-case conditions | Media fill invalid if incomplete |
Zero Contamination Expectation
For most aseptic filling operations, zero contaminated units is the expected outcome. Regulatory authorities generally consider any contaminated unit in small to moderate batch sizes to represent a process failure unless conclusively attributed to an external, assignable cause. Investigation Requirements
Any contaminated unit triggers documented investigation. The investigation must evaluate:
• Intervention timing and type
• Environmental monitoring data
• Operator activities
• Equipment performance
• Barrier integrity
• Incubation and inspection controls
Root cause must be identified and supported by objective evidence. If no assignable cause is confirmed, the media fill is considered failed and must be repeated after corrective actions.
Repeat Media Fill Criteria
Repeat simulation is required when:
• Acceptance criteria are not met
• Investigation identifies systemic risk
• Significant unmitigated deviation is detected
• Incubation or growth promotion validation fails
Acceptance criteria must be risk-based, defensible, and consistently applied. They represent the final microbiological confirmation of aseptic process control.
9. Investigation of Contaminated Units
Detection of any contaminated unit during media fill triggers a formal investigation. The objective is to determine whether contamination resulted from an identifiable, correctable cause or represents loss of aseptic process control. Investigation must be structured, documented, and initiated immediately after detection.
Initial Actions
• Segregate contaminated unit
• Confirm visual finding
• Document unit identification and location in run sequence
• Secure all related records including batch documentation and environmental monitoring data
Trend of contamination location, time, or intervention sequence must be established before proceeding to root cause hypothesis.
Intervention Timing Analysis
Intervention logs must be reviewed to determine:
• Whether contamination occurred proximal to a specific intervention
• Type of intervention performed
• Duration of exposure
• Operator performing the intervention
• Any deviation from defined technique
Video recordings, where available, should be reviewed to assess compliance with aseptic technique and exposure duration.
Environmental Monitoring Review
Environmental data must be evaluated for:
• Viable air sample excursions
• Surface contamination trends
• Glove fingertip results
• Non-viable particle spikes
• Alert or action limit exceedances
Correlation between contaminated unit position and environmental excursion timing must be assessed.
Equipment and Mechanical Assessment
Mechanical review must include:
• Needle alignment verification
• Stopper insertion consistency
• Barrier access events
• Airflow disturbance potential
• Reject system malfunction
Any unplanned equipment stoppage, adjustment, or alarm during the run must be evaluated.
Barrier and Environmental Integrity
Assessment must confirm:
• Barrier door status
• Glove integrity results
• Pressure differentials during run
• Absence of airflow interruption
Any compromise in barrier integrity requires evaluation of potential contamination pathway.
Microbiological Identification
If growth is confirmed, organism identification should be performed where possible. Identification supports determination of contamination source, such as:
• Environmental flora
• Human-associated organisms
• Water-borne organisms
• Laboratory contamination
Organism identity must be compared to environmental monitoring isolates.
Root Cause Determination
Investigation must conclude whether contamination is:
• Assignable to a specific, documented event
• Attributable to laboratory or incubation error
• Indicative of systemic aseptic process failure
Absence of a clearly supported assignable cause results in classification as process failure.
Corrective and Preventive Actions
Depending on findings, actions may include:
• Operator retraining and requalification
• Revision of intervention procedures
• Mechanical adjustment or redesign
• Environmental control enhancement
• Partial or full requalification
• Repeat media fill
Process release must not occur until investigation is complete, corrective actions are implemented, and effectiveness is verified.
A contaminated media fill is not resolved by explanation alone. It requires documented evidence, risk assessment, and corrective action sufficient to restore validated aseptic control.
10. Requalification Frequency
Media fill requalification frequency depends on:
• Production frequency
• Product type
• Regulatory commitments
• Historical performance
Periodic simulation is required to maintain validated status even in absence of major change.
11. Lifecycle Integration
Media fill is not a one-time exercise. It is part of ongoing aseptic process control. It confirms that:
• Mechanical qualification remains effective
• Environmental controls remain stable
• Personnel maintain aseptic discipline
• Process changes have not compromised sterility
Media fill therefore represents the microbiological confirmation of the aseptic filling process and provides the highest level of evidence for sterility assurance in aseptic manufacturing.