
Pharmaceutical Validation Requirements — 21 CFR Part 211
Title 21 of the Code of Federal Regulations, Part 211, commonly referred to as current Good Manufacturing Practice cGMP regulations, establishes the minimum requirements for the manufacture, processing, packing, and holding of drug products intended for human use in the United States. These regulations are issued and enforced by the U.S. Food and Drug Administration and represent a legally binding framework for pharmaceutical manufacturers.
Compliance with 21 CFR Part 211 is not discretionary. Adherence to cGMP requirements is fundamental to ensuring that drug products are safe, effective, and of consistent quality, and failures in validation are among the most common and consequential FDA inspection findings.
Regulatory Authority and Applicable Regulations
In the United States, pharmaceutical products are regulated under the Federal Food, Drug, and Cosmetic Act (FD&C Act), which provides the statutory authority for FDA oversight of drug manufacturing, testing, and distribution. FDA implements this authority through Title 21 of the Code of Federal Regulations, including 21 CFR Parts 210 and 211, which establish current Good Manufacturing Practice requirements for finished pharmaceuticals. These regulations require manufacturers to establish and maintain validated processes, qualified facilities and equipment, reliable analytical methods, and effective quality systems to ensure that drug products are safe, effective, and of consistent quality throughout their lifecycle. Compliance with these requirements is mandatory and subject to routine FDA inspection and enforcement by the U.S. Food and Drug Administration.
Role of cGMPs in Pharmaceutical Validation
The cGMP regulations provide a structured framework intended to ensure that pharmaceutical products are consistently manufactured in accordance with predefined quality standards. Validation is a core element of this framework and serves as documented evidence that processes, systems, equipment, and methods perform as intended and remain in a state of control.
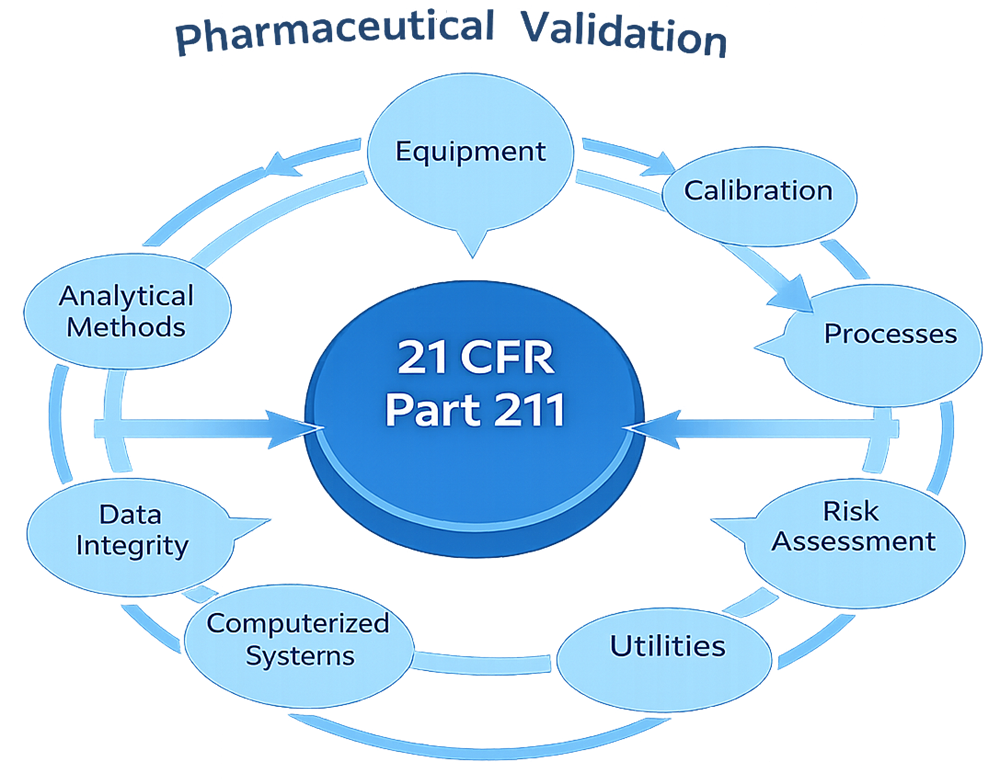
21 CFR Part 211 addresses validation expectations across multiple operational domains, including facilities, equipment, manufacturing processes, analytical methods, cleaning, utilities, computerized systems, and packaging operations. Although the regulation is not prescriptive in defining validation methodologies, FDA expects manufacturers to apply sound scientific principles, risk-based approaches, and documented lifecycle control.
Equipment Requirements — Subpart D
Subpart D — Equipment, encompassing §§ 211.63 through 211.68, establishes requirements for the design, size, construction, location, cleaning, maintenance, and calibration of manufacturing equipment.
Equipment used in pharmaceutical manufacturing must be:
- Appropriately designed for its intended use
- Constructed of suitable materials to prevent contamination
- Cleanable and maintainable
- Calibrated at defined intervals
From a validation perspective, these requirements are typically addressed through equipment qualification activities, including installation qualification, operational qualification, and where appropriate, performance qualification. Failure to demonstrate equipment suitability and control remains a frequent basis for regulatory observations.
Process Validation
Process validation is addressed in § 211.100, which requires written procedures designed to ensure that drug products possess the identity, strength, quality, and purity they purport to have.
FDA expectations for process validation are further clarified in the guidance document “Process Validation: General Principles and Practices.” This guidance establishes a lifecycle approach encompassing:
- Process design
- Process qualification
- Continued process verification
Manufacturers are expected to demonstrate that manufacturing processes are capable, reproducible, and remain in a state of control throughout routine production.
Analytical Method Validation
Although 21 CFR Part 211 does not explicitly define analytical method validation requirements in a single section, expectations are embedded throughout the regulation, particularly in requirements related to laboratory controls, testing, and data reliability.
FDA guidance documents, including “Analytical Procedures and Methods Validation for Drugs and Biologics,” provide detailed recommendations regarding method accuracy, precision, specificity, linearity, and robustness. Validated analytical methods are essential to ensuring the reliability of test results used to release and evaluate drug products.
Cleaning Validation
Cleaning validation is explicitly addressed in § 211.67, which requires that equipment be cleaned, maintained, and sanitized to prevent contamination or carryover.
Cleaning validation programs are expected to demonstrate that residues from previous products, cleaning agents, and microbial contaminants are consistently reduced to acceptable levels. FDA guidance and industry practice emphasize:
- Defined acceptance criteria
- Worst-case product selection
- Validated sampling and analytical methods
Inadequate cleaning validation continues to be a common source of FDA inspection findings.
Computerized Systems and Data Integrity
While computer system validation is not explicitly named in 21 CFR Part 211, requirements related to data integrity, record accuracy, and control of electronic systems are implicit throughout the regulation.
FDA guidance on electronic records and electronic signatures provides expectations for validation of computerized systems that create, modify, maintain, or archive GMP data. Systems that impact product quality, patient safety, or regulatory decisions must be validated and maintained in a controlled state.
Validation of Utilities
21 CFR Part 211 does not explicitly prescribe validation requirements for utilities; however, utilities such as purified water, water for injection, clean steam, compressed gases, and HVAC systems are recognized as critical to product quality.
FDA inspectors routinely expect documented evidence that critical utilities are designed, qualified, and monitored appropriately. Industry guidance, including ISPE baseline guides, is commonly used to support defensible utility validation programs.
Packaging and Container Closure Validation
While packaging validation is not explicitly detailed in Part 211, requirements related to packaging, labeling, and container closure systems are addressed throughout the regulation.
FDA guidance on container closure systems for human drugs and biologics outlines expectations for demonstrating that packaging components protect the drug product throughout its shelf life. Packaging validation activities typically address compatibility, integrity, and protection against environmental factors.
Regulatory Perspective
Validation under 21 CFR Part 211 is not a one-time exercise. FDA expects pharmaceutical manufacturers to maintain validated systems through effective change control, deviation management, and ongoing monitoring. Validation deficiencies are often interpreted as systemic quality failures rather than isolated technical gaps.
Bottom Line
21 CFR Part 211 establishes the regulatory foundation for pharmaceutical manufacturing in the United States. Validation is a central pillar of cGMP compliance and a primary focus of FDA inspections. Manufacturers that apply conservative, lifecycle-based validation practices and maintain disciplined documentation are best positioned to demonstrate sustained compliance and inspection readiness.