
Sterile Component Preparation and Transfer
Sterility assurance in aseptic filling does not begin at the filling needle. It begins with the preparation, sterilization, protection, and controlled transfer of all components that will contact sterile product or enter the ISO 5 critical exposure zone. A failure in component control cannot be corrected during filling. Therefore, sterile component management must be treated as a lifecycle-controlled process integrated into overall aseptic strategy.
This article defines how components are prepared, sterilized or depyrogenated, protected from recontamination, and transferred into the aseptic environment while maintaining validated sterility assurance.
1. Scope and Risk Classification
Sterile components include all materials that directly contact sterile product or are present within the ISO 5 exposure zone. These typically include glass vials, syringes, elastomeric stoppers, aluminum seals, product-contact tubing, filling needles, sterile filters, and reusable tools introduced into the critical zone.
The diagram below illustrates the sterile component lifecycle from initial preparation through sterilization, protected storage, controlled transfer, and final presentation to the aseptic filling line. It emphasizes that sterility assurance is a continuous chain of controlled steps rather than a single sterilization event.
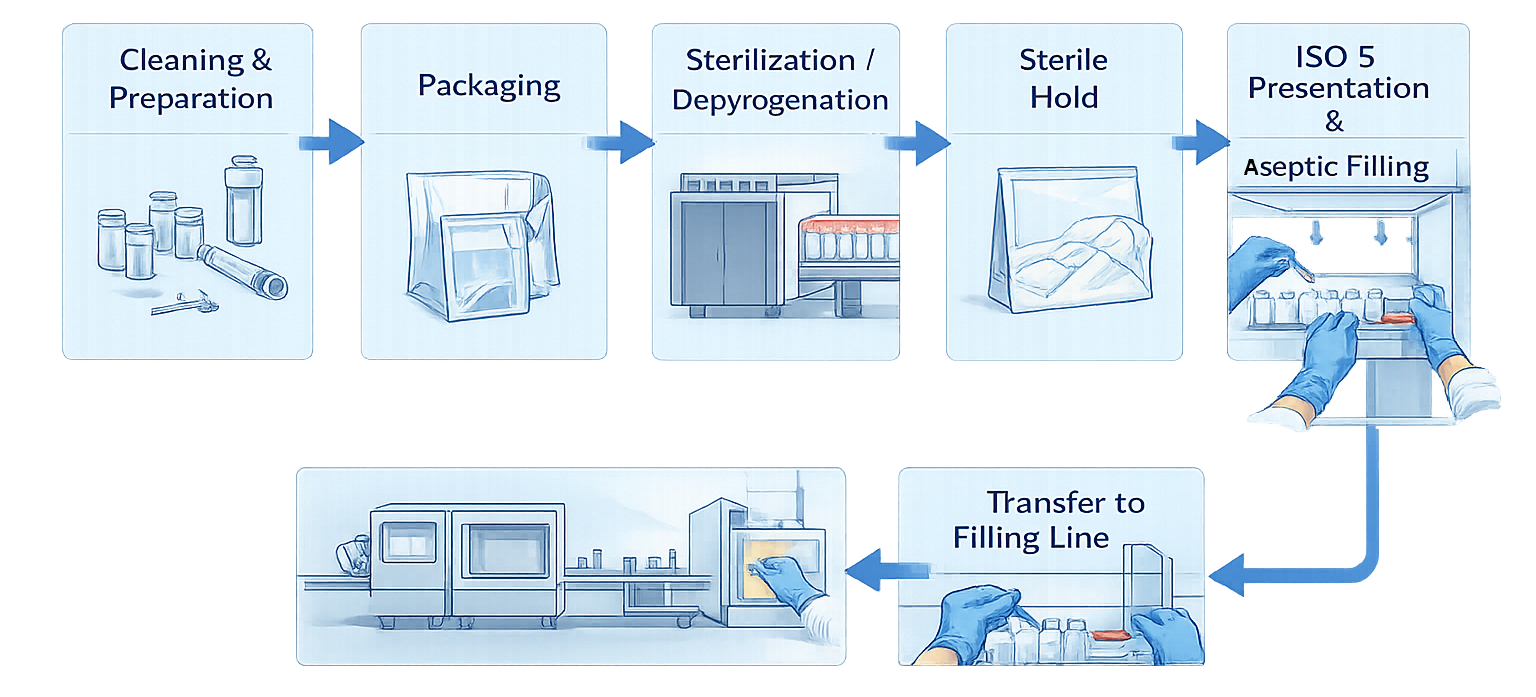
Not all components carry identical risk. Risk classification must consider:
- Direct product contact
- Duration of exposure in ISO 5
- Potential for endotoxin contamination
- Complexity of surface geometry
- Reusability versus single-use
Product-contact components represent the highest contamination risk. Closure components such as stoppers and caps are critical to container integrity and sterility maintenance. Accessories entering ISO 5 without direct product contact still require controlled handling because they may become secondary contamination sources.
Risk classification drives sterilization method selection, validation depth, environmental staging controls, and transfer procedures.
2. Cleaning and Pre-Sterilization Preparation
Reusable components require validated cleaning prior to sterilization. Cleaning validation must demonstrate effective removal of:
- Product residues
- Cleaning agents
- Microbial contamination
- Endotoxin where applicable
Validation must define worst-case soils, cleaning parameters, and sampling methods. Components must be visually inspected for integrity and cleanliness before packaging for sterilization. Damaged components must be rejected.
Packaging configuration is part of the sterilization design. Packaging must allow sterilant penetration while protecting surfaces from post-cycle contamination. Overpacking or incorrect wrapping can compromise sterilization effectiveness.
For single-use components, the control model shifts toward supplier oversight. Manufacturers must qualify suppliers, verify sterilization method and sterility assurance level, confirm packaging integrity, and ensure traceability to sterilization lot documentation. Single-use does not eliminate validation responsibility; it transfers part of the control to supplier qualification and incoming verification.
3. Sterilization and Depyrogenation Methods
Sterilization method selection must be scientifically justified based on material compatibility and contamination risk.
Dry heat depyrogenation is commonly used for glass vials and syringes. It provides both sterilization and endotoxin destruction. Validation requires temperature mapping across the tunnel, verification of conveyor speed, and endotoxin challenge demonstrating required log reduction. Depyrogenation is critical for injectable products because endotoxin cannot be removed by sterile filtration.
Steam sterilization is typically used for reusable product-contact components. Validation must define load configuration, worst-case positioning, biological indicator placement, and heat penetration performance. Cycle parameters must demonstrate consistent lethality under defined worst-case conditions.
Gamma irradiation is commonly used for single-use assemblies. Manufacturers must verify supplier dose validation and sterility assurance level. Incoming inspection must confirm packaging integrity and lot traceability.
Vaporized hydrogen peroxide may be used for barrier interior components or isolator-based transfer systems. Validation must demonstrate uniform distribution, biological indicator inactivation, and acceptable residual levels.
Each sterilization method requires documented qualification and ongoing periodic requalification.
4. Post-Sterilization Protection and Hold Time
Sterility achieved during sterilization must be preserved until use. Post-sterilization controls include defined sterile hold times, controlled storage conditions, and protection from physical damage.
Sterile hold time must be justified by data. Environmental classification of staging areas must be appropriate to the risk level of the component. Packaging integrity must remain intact until transfer. Excessive hold times without validation undermine sterility assurance.
5. Transfer into ISO 5
Transfer of sterilized components into the ISO 5 critical exposure zone represents one of the highest contamination risk stages in the sterile component lifecycle. Sterilization alone does not guarantee sterility at the point of use. Sterility must be preserved through controlled handling, protected staging, and validated transfer procedures.
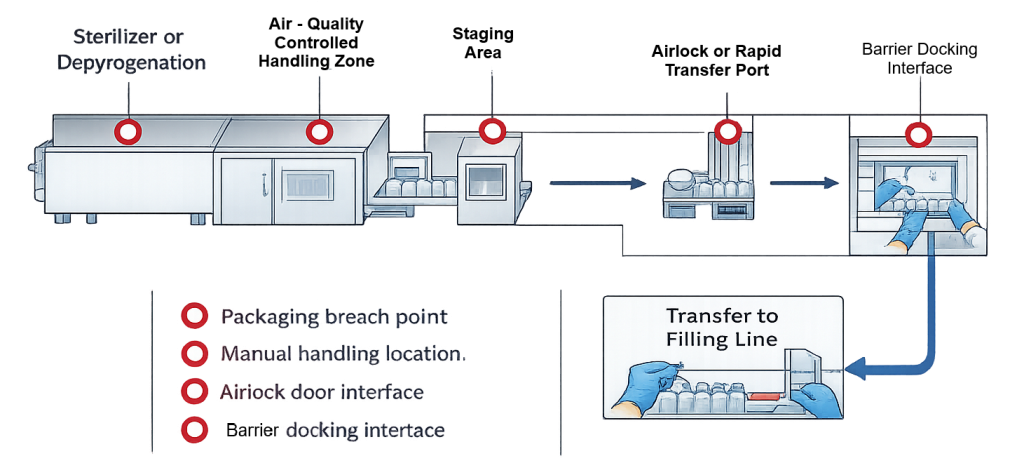
The transfer process typically includes removal from the sterilizer or depyrogenation tunnel, staging in a controlled environment, passage through an airlock or rapid transfer port, and final introduction into the ISO 5 filling zone. Each interface in this sequence presents a potential contamination pathway. Primary risk points include:
• Breach of the primary sterile barrier during packaging removal
• Manual manipulation of sterilized components
• Airlock door interfaces and pressure transitions
• Barrier docking interfaces
• Stopper bowl or component loading points
Automated transfer systems, such as direct tunnel-to-filler connections or closed conveyor interfaces, reduce manual exposure but still require verification of mechanical alignment, enclosure integrity, and airflow compatibility. When manual transfer is required, procedures must define:
• Environmental classification of staging and unwrapping zones
• Controlled removal of outer packaging
• Preservation of inner sterile barrier until ISO 5 entry
• Minimization of exposure duration
• Operator qualification and aseptic technique requirements
Transfer procedures must be validated, documented, and periodically reviewed. Environmental monitoring should support verification that transfer operations do not compromise ISO 5 conditions.
Sterility is preserved only when the entire transfer chain remains controlled. The risk map above visually identifies the locations where loss of control most commonly occurs and where procedural discipline must be strongest.
6. Presentation to the Filling Line
Once inside ISO 5, components must be presented in a manner that preserves first air protection. Stopper bowls, cap hoppers, and feed tracks must be loaded without disrupting airflow or contacting non-sterile surfaces.
Mechanical systems must prevent excessive vibration, component abrasion, or pile-up that may generate particles. Improper presentation can negate validated sterilization controls.
7. Intervention Risk During Component Handling
Component replenishment and adjustment represent contamination risks. Stopper bowl loading, cap hopper filling, and manual tool introduction must be assessed during media fill and environmental monitoring studies.
Interventions must be predefined and incorporated into aseptic process simulation. High-frequency or high-exposure interventions require additional procedural control and operator training.
8. Validation and Documentation
Sterile component lifecycle control requires comprehensive documentation, including:
- Cleaning validation reports
- Sterilization and depyrogenation qualification protocols
- Biological indicator data
- Load configuration justification
- Sterile hold time studies
- Transfer validation documentation
Traceability must link each component lot to sterilization cycle and batch record.
9. Change Management and Requalification
Changes that may impact sterility include:
- Modification of load configuration
- Change in sterilization cycle parameters
- Introduction of new component materials
- Supplier changes
- Modification of transfer methods
Each change must undergo documented risk assessment. Requalification depth must be proportional to sterility impact.
10. Lifecycle Control
Sterile component preparation and transfer must remain under continued verification. This includes periodic sterilizer requalification, supplier performance review, environmental monitoring trend analysis, and audit of transfer procedures.
Sterility assurance begins before filling and must be preserved until final container closure. Aseptic filling cannot compensate for inadequate preparation, sterilization, or transfer of components.