
Steam Sterilization Qualification Lifecycle
Steam sterilization qualification demonstrates that a sterilization system consistently delivers the required level of microbial lethality under defined and controlled operating conditions. Qualification is not a single event but a structured lifecycle process designed to establish, document, and confirm sterilization capability.
The lifecycle approach ensures that equipment performance, thermal distribution, load penetration, and microbiological lethality are evaluated in a logical and reproducible manner. Each phase of qualification builds upon the previous one, forming a defensible foundation for routine sterilization operations.
Regulatory and Standards Framework
Steam sterilization qualification aligns with international regulatory and industry standards, including:
- 21 CFR 211.113 – Control of microbiological contamination
- EU GMP Annex 1 – Sterilization process validation
- ISO 17665 – Moist heat sterilization validation
- EN 285 – Large steam sterilizers and steam quality requirements
- PDA Technical Report No. 1 – Steam sterilization process design and validation
These standards define expectations for:
- Process development
- Heat distribution studies
- Biological indicator use
- Steam quality
- Documentation and lifecycle control
The steam sterilization qualification process follows a structured lifecycle, progressing from system definition through qualification and ongoing control. The following diagram illustrates the sequence and interrelationship of each phase.
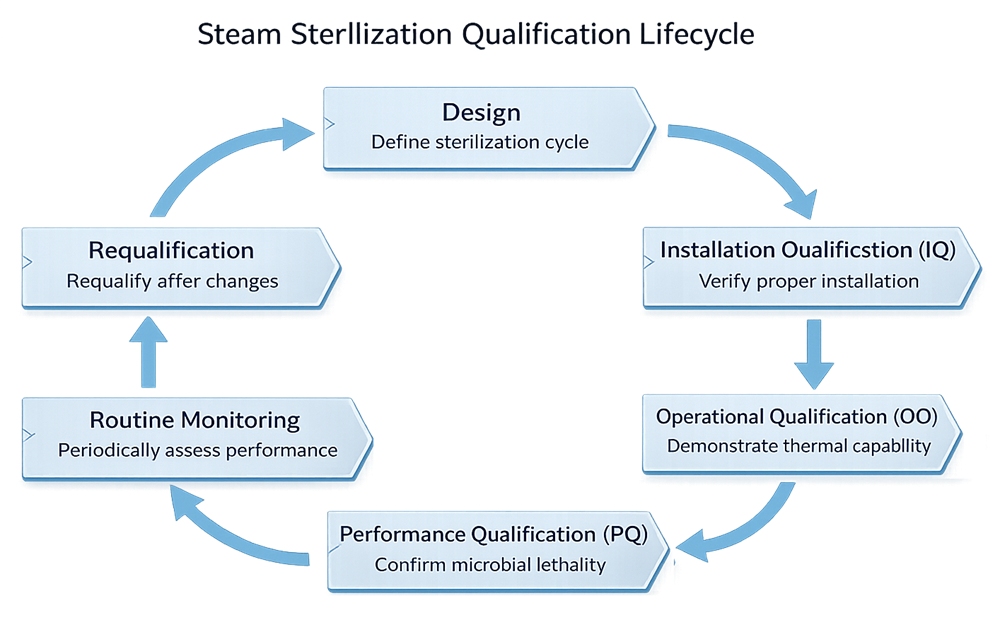
1. Installation Qualification
Installation Qualification verifies that the sterilizer and supporting utilities are installed in accordance with approved specifications.
Regulatory Context
- 21 CFR 211.63 requires equipment to be of appropriate design and size.
- 21 CFR 211.68 requires appropriate controls over automatic equipment.
- ISO 17665 requires documented installation verification prior to operational testing.
Key Elements
- Equipment identification and configuration
- Utility verification
- Steam supply qualification
- Instrument installation and calibration
- Control system configuration
- Safety interlocks
IQ confirms that the system is correctly installed and ready for operational testing.
2. Operational Qualification
Operational Qualification demonstrates that the sterilizer operates within defined parameters and achieves required thermal performance.

Temperature mapping system connected to steam sterilizer during qualification study
Regulatory Context
- 21 CFR 211.113 requires validated sterilization processes.
- EU Annex 1 requires validation of sterilization cycles prior to use.
- ISO 17665 specifies heat distribution and air removal verification.
Core OQ Elements
Control System Verification
- Cycle sequencing
- Alarm functionality
- Interlocks
- Data recording integrity
Vacuum Integrity
- Leak rate testing
- Air removal verification
- Bowie-Dick testing for pre-vacuum systems
Empty Chamber Heat Distribution
- Temperature mapping
- Cold spot identification
- Drain monitoring
Operational Qualification establishes that the sterilizer performs as intended under empty chamber conditions.
3. Performance Qualification
Performance Qualification confirms that the sterilization cycle achieves required lethality under defined worst-case load conditions.
Regulatory Context
- ISO 17665 requires demonstration of lethality under defined loading patterns.
- EU Annex 1 requires validation under worst-case conditions.
- FDA expects reproducibility and documented evidence of microbial inactivation.
PQ Elements
Loaded Heat Penetration
- Worst-case probe placement
- Confirmation of slowest-heating location
- Exposure verification
Biological Indicators
- Placement at worst-case penetration points
- Use of appropriate challenge organism
- Documented inactivation
Reproducibility
- Typically three consecutive successful runs
The sterilization cycle is validated at the slowest-heating location.
4. Lethality Determination
Lethality is expressed as F₀, calculated at the worst-case probe location.
ISO 17665 requires that:
- Lethality calculations are documented
- Acceptance criteria are predefined
- Deviations are evaluated
All probes must achieve required exposure conditions.
5. Documentation and Approval
Regulatory expectation requires:
- Approved validation protocol
- Traceable raw data
- Calibration documentation
- Deviation investigation
- Biological indicator results
- Summary report with technical justification
Per 21 CFR 211.194, laboratory records must be complete and attributable.
The sterilizer is considered qualified only after formal approval of the validation report.
6. Lifecycle Control and Continued Compliance
Regulatory authorities expect sterilization processes to remain in a validated state.
EU Annex 1 and ISO 17665 both require:
- Ongoing monitoring
- Change impact assessment
- Periodic review
- Requalification when necessary
Qualification establishes capability.
Lifecycle control preserves compliance.