
VHP Sterilization Validation and Lifecycle Control
1. Introduction
VHP sterilization validation demonstrates that a vapor hydrogen peroxide system consistently achieves the required level of microbial inactivation under defined operating conditions. Validation is not limited to initial qualification. It is a lifecycle-controlled activity that includes:
- Defined validation strategy
- Structured qualification execution
- Routine performance monitoring
- Change control integration
- Periodic review and requalification
In regulated pharmaceutical environments, VHP sterilization is treated as a critical process requiring documented scientific justification and ongoing control.
2. Regulatory and Industry Framework
VHP sterilization validation in pharmaceutical manufacturing operates within an established regulatory framework. Regulatory authorities do not prescribe specific cycle parameters. Instead, they require scientific validation, documented evidence, and lifecycle control demonstrating consistent microbial inactivation performance.
2.1 U.S. Regulatory Expectations
Under U.S. current Good Manufacturing Practice requirements:
- 21 CFR 211.63 requires equipment to be of appropriate design and suitable for intended use.
- 21 CFR 211.67 requires cleaning and maintenance of equipment.
- 21 CFR 211.113(b) requires written procedures designed to prevent microbiological contamination of sterile drug products.
- 21 CFR 211.68 addresses automatic, mechanical, and electronic equipment controls.
Although VHP is not explicitly named in the regulation, systems used to support aseptic processing fall under these requirements.
For outsourcing facilities operating under Section 503B of the FD&C Act, expectations for sterility assurance and environmental control are equally applicable.
2.2 International and Industry Guidance
In addition to U.S. regulations, the following guidance documents influence VHP validation expectations:
- EU GMP Annex 1 Manufacture of Sterile Medicinal Products
- ISO 14937 Sterilization of health care products – General requirements for characterization of a sterilizing agent and development, validation, and routine control of a sterilization process
- PDA Technical Reports addressing isolator and bio-decontamination technologies
- ICH Q9 Quality Risk Management
These documents reinforce:
- Risk-based validation depth
- Defined sterility assurance objectives
- Control of critical parameters
- Lifecycle performance monitoring
2.3 Regulatory Focus Areas During Inspection
Regulatory inspections typically evaluate:
- Scientific justification of validation approach
- Worst-case challenge rationale
- Biological indicator selection and placement logic
- Definition and control of critical parameters
- Change control linkage
- Periodic performance review
Inspectors assess not only whether validation was performed, but whether it is scientifically justified and maintained under control.
3. Validation Strategy and Intended Use
Validation scope must reflect intended application. VHP systems are commonly used for:
- Isolator decontamination
- RABS decontamination
- Pass-through chamber sterilization
- Room bio-decontamination
The validation strategy must define:
- Required sterility assurance objective
- Target log reduction
- Worst-case load configuration
- Critical process parameters
- Acceptance criteria
Validation depth is risk-based and aligned with system impact on product sterility.
4. Lifecycle Approach to VHP Validation
VHP validation follows a structured lifecycle consistent with GMP expectations.
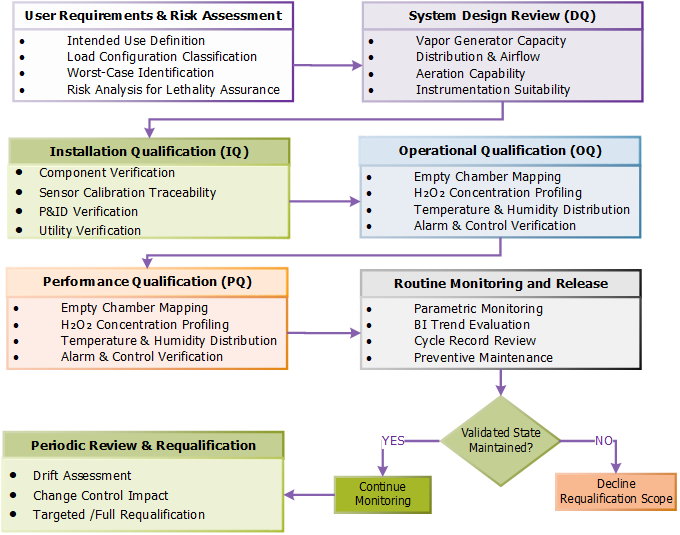
4.1 Design Qualification (if applicable)
- System suitability for intended use
- Airflow modeling or distribution evaluation
- Material compatibility review
- Control system capability
For integrated isolators, DQ may be part of supplier documentation review.
4.2 Installation Qualification (IQ)
IQ confirms:
- Equipment installed per specifications
- Utility connections verified
- Component identification and traceability
- Instrument calibration status
- Safety interlocks functional
Documentation review and physical verification are essential.
4.3 Operational Qualification (OQ)
OQ verifies that the system operates within defined parameter ranges. Typical OQ elements:
- Empty chamber mapping
- Temperature mapping
- Humidity behavior verification
- Concentration profile assessment
- Alarm verification
- Cycle parameter repeatability
OQ establishes parameter control capability before biological challenge.
4.4 Performance Qualification (PQ)
Performance Qualification demonstrates that the VHP sterilization process consistently achieves the required microbiological lethality under defined worst-case operating conditions.
PQ must be conducted using scientifically justified challenge conditions representative of routine production.
Key PQ Elements
- Biological indicator placement strategy
- Worst-case load configuration
- Replicate qualification runs
- Target log reduction confirmation
- Predefined acceptance criteria
PQ execution must reflect realistic operational loading patterns, material configurations, and environmental conditions.
Worst-Case Load Configuration Concept
Worst-case configuration is a critical element of VHP performance qualification. The load selected for PQ must represent the most difficult-to-sterilize condition based on documented risk assessment.
Worst-case conditions may be driven by:
- Vapor penetration limitations
- Diffusion restrictions
- Load density and material mass
- Restricted lumens or enclosed geometries
- Airflow shadowing and vapor distribution patterns
The illustration below demonstrates typical worst-case characteristics, including high-density loading, nested components, restricted lumens, obstructed vapor paths, and biological indicator placement in areas expected to receive the lowest hydrogen peroxide concentration. Worst-case selection must be documented, risk-justified, and traceable to the PQ protocol.
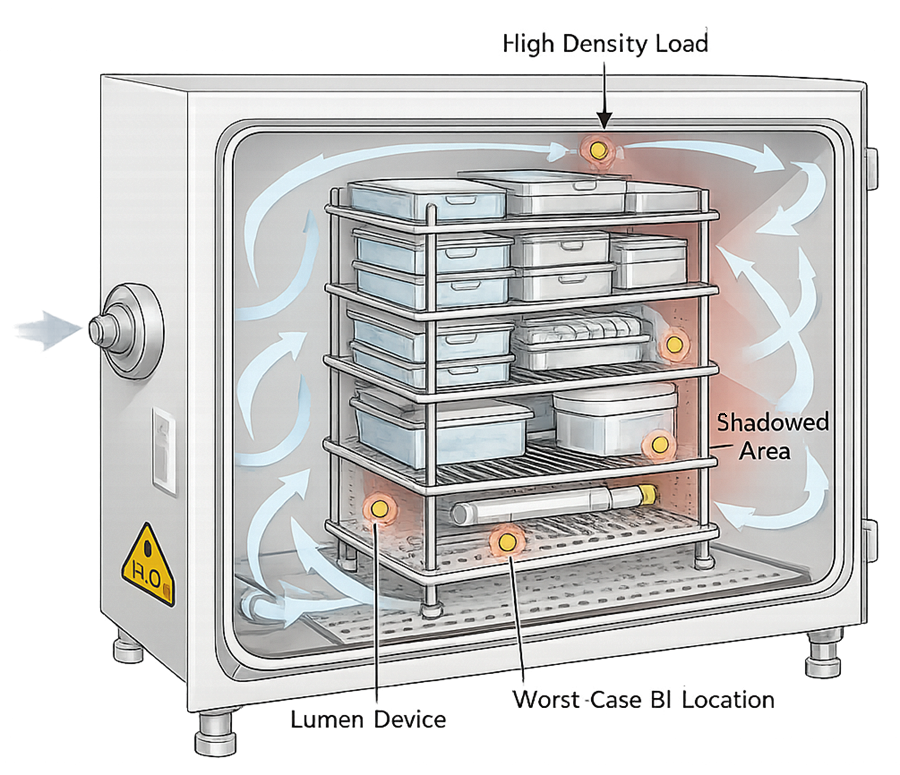
Representative examples of high-density load configurations evaluated during VHP performance qualification.


5. Biological Indicator Strategy
Biological indicators are used during Performance Qualification to directly challenge the microbiological lethality of the VHP sterilization process.
For VHP sterilization, BIs typically contain Geobacillus stearothermophilus spores due to their demonstrated resistance to hydrogen peroxide vapor and established D-value characteristics. BI population and resistance specifications must be appropriate for the required sterility assurance level and supported by supplier certification. BI lot qualification and certificate review shall confirm:
- Certified spore population
- D-value under defined conditions
- Expiration status
- Storage requirements
BI Placement Strategy
Biological indicators are placed at locations representing the most difficult-to-sterilize conditions within the chamber, including:
- Areas of lowest airflow velocity
- Shadowed regions
- Downstream of obstructions
- Furthest points from vapor injection
- Within restricted lumens or enclosed geometries
Placement must be justified based on:
- Documented airflow distribution
- Load geometry and material density
- Vapor penetration limitations
- Formal risk assessment
BI placement must never be arbitrary. Locations shall be predefined in the PQ protocol and traceable to the worst-case load configuration rationale.
Incubation and Result Interpretation
Following exposure, BIs must be incubated under manufacturer-specified conditions to ensure reliable recovery of surviving organisms. Incubation parameters shall include:
- Defined temperature range
- Minimum incubation duration
- Positive control verification
- Negative control verification
Incubation conditions must be documented and monitored. Early readout systems, if used, must be validated and supported by manufacturer data.
A successful PQ run requires complete inactivation of all exposed BIs while demonstrating growth in positive controls.
6. Critical Process Parameters
VHP sterilization effectiveness depends on precise control of defined critical process parameters. These parameters directly influence vapor distribution, microbial lethality, material compatibility, and residual removal. Typical critical parameters include:
- VHP concentration or injection rate
- Exposure time
- Relative humidity
- Temperature
- Aeration duration
Each parameter contributes to overall lethality performance and must be evaluated during Operational Qualification and confirmed during Performance Qualification.
VHP Concentration or Injection Rate
Hydrogen peroxide concentration directly influences sporicidal activity. Insufficient vapor concentration may result in sublethal exposure, while excessive concentration may cause material compatibility concerns or condensation. Injection rate must ensure:
- Rapid achievement of target concentration
- Uniform distribution
- Avoidance of liquid-phase condensation unless specifically intended
Concentration limits shall be established based on development studies, mapping data, and microbiological challenge results.
Exposure Time
Exposure duration determines cumulative lethality delivered to biological indicators and product surfaces. Minimum exposure time must be validated to demonstrate required log reduction under worst-case load conditions. Time control shall account for:
- Ramp-up phase
- Dwell phase
- Stabilization variability
Exposure limits must be predefined and verified through repeatability during PQ.
Relative Humidity
Humidity significantly affects hydrogen peroxide efficacy. Adequate moisture enhances spore susceptibility, while excessively low humidity may reduce lethality. Humidity control must ensure:
- Stable preconditioning conditions
- Uniform distribution throughout the chamber
- Reproducibility across runs
Humidity setpoints shall be supported by lethality data generated during development or OQ.
Temperature
Temperature influences both vapor behavior and microbial resistance characteristics. While VHP processes are typically low-temperature sterilization methods, temperature stability remains critical for:
- Vapor saturation behavior
- Prevention of condensation
- Consistent D-value performance
Temperature mapping during OQ must confirm uniformity within validated limits.
Aeration Duration
Aeration ensures removal of residual hydrogen peroxide following exposure. Inadequate aeration may lead to:
- Residual chemical risk
- Material compatibility concerns
- Occupational safety issues
Aeration time must be validated to demonstrate acceptable residual levels where applicable.
Establishment of Parameter Limits
Critical parameter limits must be:
- Scientifically justified
- Defined and approved prior to execution
- Traceable to development data and validation rationale
- Controlled through validated instrumentation and alarm systems
Acceptance ranges shall reflect worst-case qualification results and incorporate appropriate safety margins without unnecessarily constraining routine operation. Parameter limits must not be adjusted post-execution to accommodate failing results. Any modification requires documented investigation and revalidation assessment.
Acceptance Criteria
Acceptance criteria typically include:
- Required log reduction of biological indicator
- Defined exposure time at target concentration
- Maximum allowable parameter deviations
- Successful aeration to safe re-entry levels
Acceptance criteria must be established before validation execution.
7. Requalification and Continued Verification
Requalification is required whenever changes or performance signals indicate potential impact to the validated state of the VHP sterilization process. Triggers for requalification may include:
- Major maintenance activities affecting critical components
- Control system modification or software updates
- Chamber integrity repair or structural intervention
- Load configuration changes
- Repeated deviation trends or adverse performance signals
All requalification decisions must be supported by documented impact assessment and risk evaluation.
Scope and Depth of Requalification
The extent of requalification shall be commensurate with the nature of the change and the assessed risk to sterilization lethality. Requalification activities may include:
- Targeted verification testing of affected parameters
- Partial Performance Qualification under defined worst-case conditions
- Full requalification including repeat PQ execution
The scope must be predefined, justified, and approved prior to execution.
Periodic Review and Continued Verification
Continued process suitability must be confirmed through structured periodic review of performance data. Periodic review shall assess:
- Stability and consistency of critical parameter trends
- Frequency and significance of deviations
- Alarm events and control system alerts
- Impact of maintenance interventions
- Evidence of performance drift
Periodic review is not a retrospective exercise. It is an active lifecycle control mechanism used to confirm continued fitness for intended use and determine whether requalification is required. Lifecycle control ensures that validation remains a maintained state rather than a historical event.
8. Documentation and Data Integrity
Validation documentation must include:
- Approved protocol
- Executed data
- Deviations and investigations
- Final report with conclusion
- Change control linkage
Electronic data must comply with data integrity expectations where applicable.